Advanced Biologic Therapies and Advanced Biologics Manufacturing
Crowell LE, Lu AE, Love KR, Stockdale A, Timmick SM, Wu D, Wang YA, Doherty W, Bonnyman A, Vecchiarello N, Goodwine C, Bradbury L, Brady JR, Clark JJ, Colant NA, Cvetkovic A, Dalvie NC, Liu D, Liu Y, Mascarenhas CA, Matthews CB, Mozdzierz NJ, Shah KA, Wu SL, Hancock WS, Braatz RD, Cramer SM, Love JC.On-demand manufacturing of clinical-quality biopharmaceuticals.Nat Biotech2018;36:988–995.
Abstract: Conventional manufacturing of protein biopharmaceuticals in centralized, large-scale, single-product facilities is not well-suited to the agile production of drugs for small patient populations or individuals. Previous solutions for small-scale manufacturing are limited in both process reproducibility and product quality, owing to their complicated means of protein expression and purification. We describe an automated, benchtop, multiproduct manufacturing system, called Integrated Scalable Cyto-Technology (InSCyT), for the end-to-end production of hundreds to thousands of doses of clinical-quality protein biologics in about 3 d. Unlike previous systems, InSCyT includes fully integrated modules for sustained production, efficient purification without the use of affinity tags, and formulation to a final dosage form of recombinant biopharmaceuticals. We demonstrate that InSCyT can accelerate process development from sequence to purified drug in 12 weeks. We used integrated design to produce human growth hormone, interferon α-2b and granulocyte colony-stimulating factor with highly similar processes on this system and show that their purity and potency are comparable to those of marketed reference products.
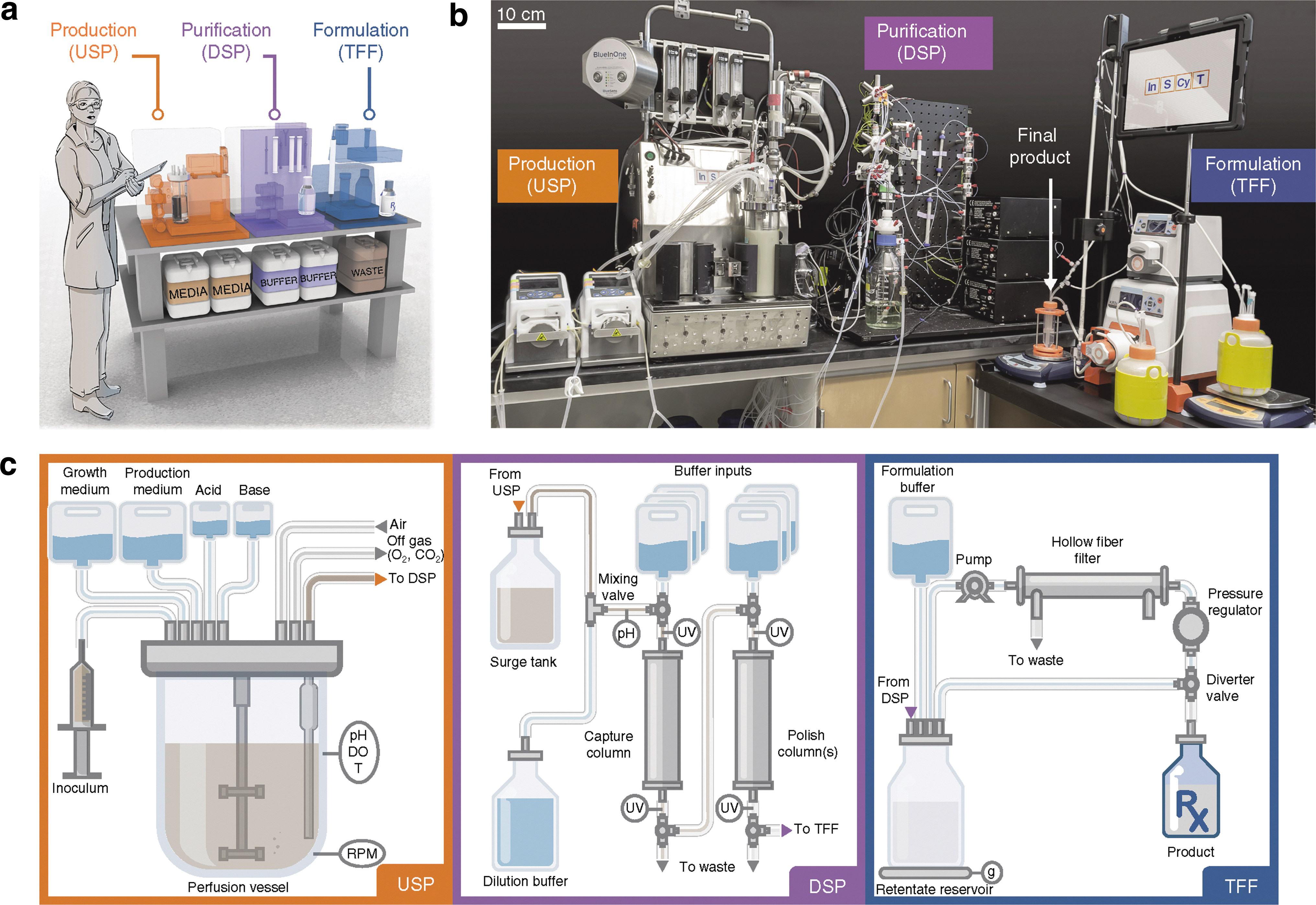
Schematic of the InSCyT system for on-demand biomanufacturing and demonstration of consistent operation across three distinct InSCyT systems. (a) To-scale rendering of the InSCyT system. Human figure is approximately 5 feet 7 inches (170 cm) tall. (b) Photograph of an operational InSCyT system. (c) Detailed schematic of the InSCyT system, including interactions between modules and key control points for the production (upstream processing, USP), purification (downstream processing, DSP) and formulation (tangential flow filtration, TFF) modules. DO, dissolved oxygen; T, temperature. (d,e) Process parameter profiles collected by the control software from the production (USP) module (d) and the purification (DSP) module (e) of three separate InSCyT systems during hGH fermentation. SLPM, standard liters per minute.
Isabella VM, Ha BN, Castillo MJ, Lubkowicz DJ, Rowe SE, Millet YA, Anderson CL, Li N, Fisher AB, West KA, Reeder PJ, Momin MM, Bergeron CG, Guilmain SE, Miller PF, Kurtz CB, Falb D.Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria.Nat Biotechnol2018;36:857–864.
Abstract: Phenylketonuria (PKU) is a genetic disease that is characterized by an inability to metabolize phenylalanine (Phe), which can result in neurotoxicity. To provide a potential alternative to a protein-restricted diet, we engineered Escherichia coli Nissle to express genes encoding Phe-metabolizing enzymes in response to anoxic conditions in the mammalian gut. Administration of oursynthetic strain, SYNB1618, to the Pahenu2/enu2 PKU mouse model reduced blood Phe concentration by 38% compared with the control, independent of dietary protein intake. In healthy Cynomolgus monkeys, we found that SYNB1618 inhibited increases in serum Phe after an oral Phe dietary challenge. In mice and primates, Phe was converted to trans-cinnamate by SYNB1618, quantitatively metabolized by the host to hippurate and excreted in the urine, acting as a predictive biomarker for strain activity. SYNB1618 was detectable in murine or primate feces after a single oral dose, permitting the evaluation of pharmacodynamic properties. Our results define a strategy for translation of live bacterial therapeutics to treat metabolic disorders.
Commentary:Two recent reports mark important milestones in the area of biologics development. The work by Crowell et al. provides a much-needed improvement in the ability to manufacture biopharmaceuticals in a modular benchtop fashion. Such a customizable production addresses a recently growing bottleneck, as many existing therapeutics are needed in new doses and formulations in order to enable exploratory personalized-medicine type trials, as well as there being a growing need to produce biologics at sites where transportation of refrigerated materials is problematic. The team built a fully automated and integrated system of protein expression vessels combined with purification and formulation modules (
first
figure
). The concept for the different units came from standard biomanufacturing, except that everything was scaled down. The team used yeast as the expression system due to its ease of genetic manipulation and the production of fewer potentially contaminating proteins. Proof of concept was demonstrated with human growth hormone, interferon α, and granulocyte colony-stimulating factor. However, Good Manufacturing Practice compatibility of this new approach has yet to be shown.
In turn, Isabella et al. present a long sought-after therapy for phenylketonuria (PKU), a genetic metabolic disease associated with inability to metabolize phenylalanine, which utilizes a live bacterial cocktail and which has the promise to eliminate the need for PKU patients to follow a protein-restricted diet. As a starting point, the group used Escherichia coli strain Nissle due to its well established safety profile and previous uses to treat inflammatory bowel disease and irritable bowel syndrome. Careful genetic engineering was required in order to provide the operon for degradation of phenylalanine, while also providing a transporter to allow phenylalanine to enter the bacterial cell for said processing (
second figure
). The team also had to tackle the reduced viability of the re-engineered bacteria by optimizing various promoters. The new strain showed a good pharmacokinetic profile, with rapid clearance, and solid data were obtained in a proof-of-concept PKU treatment in mice. Pilot studies were also run in cynomolgous monkey, paving the way to graduating the candidate therapy to human testing (initial assessments in humans under clinical study NCT03516487). Contributed by Anton Simeonov.
Engineering, activity and pharmacokinetic profiling of SYNB1618: creation of a candidate therapeutic strain. (a) SYNB1618 contains chromosomally inserted genes encoding PheP, a high-affinity Phe transporter that can bring Phe into the cell, PAL (stlA), which converts Phe into TCA, and LAAD (pma), which converts Phe to PP. Regulation of these components is carried out by anaerobic-, IPTG- and l-arabinose-inducible promoters to enable activation in the mammalian gut or in vitro.
PROTAC Reporter Assays
Riching KM, Mahan S, Corona CR, McDougall M, Vasta JD, Robers MB, Urh M, Daniel DL. Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action.ACS Chem Biol2018;13:2758–2770.
Abstract: A new generation of heterobifunctional small molecules, termed proteolysis targeting chimeras (PROTACs), targets proteins for degradation through recruitment to E3 ligases and holds significant therapeutic potential. Despite numerous successful examples, PROTAC small molecule development remains laborious and unpredictable, involving testing compounds for endpoint degradation activity at fixed times and concentrations without resolving or optimizing for the important biological steps required for the process. Given the complexity of the ubiquitin proteasomal pathway, technologies that enable real-time characterization of PROTAC efficacy and mechanism of action are critical for accelerating compound development, profiling, and improving guidance of chemical structure–activity relationship. Here, we present an innovative, modular live-cell platform utilizing endogenous tagging technologies and apply it to monitoring PROTAC-mediated degradation of the bromodomain and extraterminal family members. We show comprehensive real-time degradation and recovery profiles for each target, precisely quantifying degradation rates, maximal levels of degradation (Dmax), and time frame at Dmax. These degradation metrics show specific PROTAC and family member-dependent responses that are closely associated with the key cellular protein interactions required for the process. Kinetic studies show cellular ternary complex stability influences potency and degradation efficacy. Meanwhile, the level of ubiquitination is highly correlated to degradation rate, indicating ubiquitination stemming from productive ternary complex formation is the main driver of the degradation rate. The approaches applied here highlight the steps at which the choice of E3 ligase handle can elicit different outcomes and discern individual parameters required for degradation, ultimately enabling chemical design strategies and rank ordering of potential therapeutic compounds.
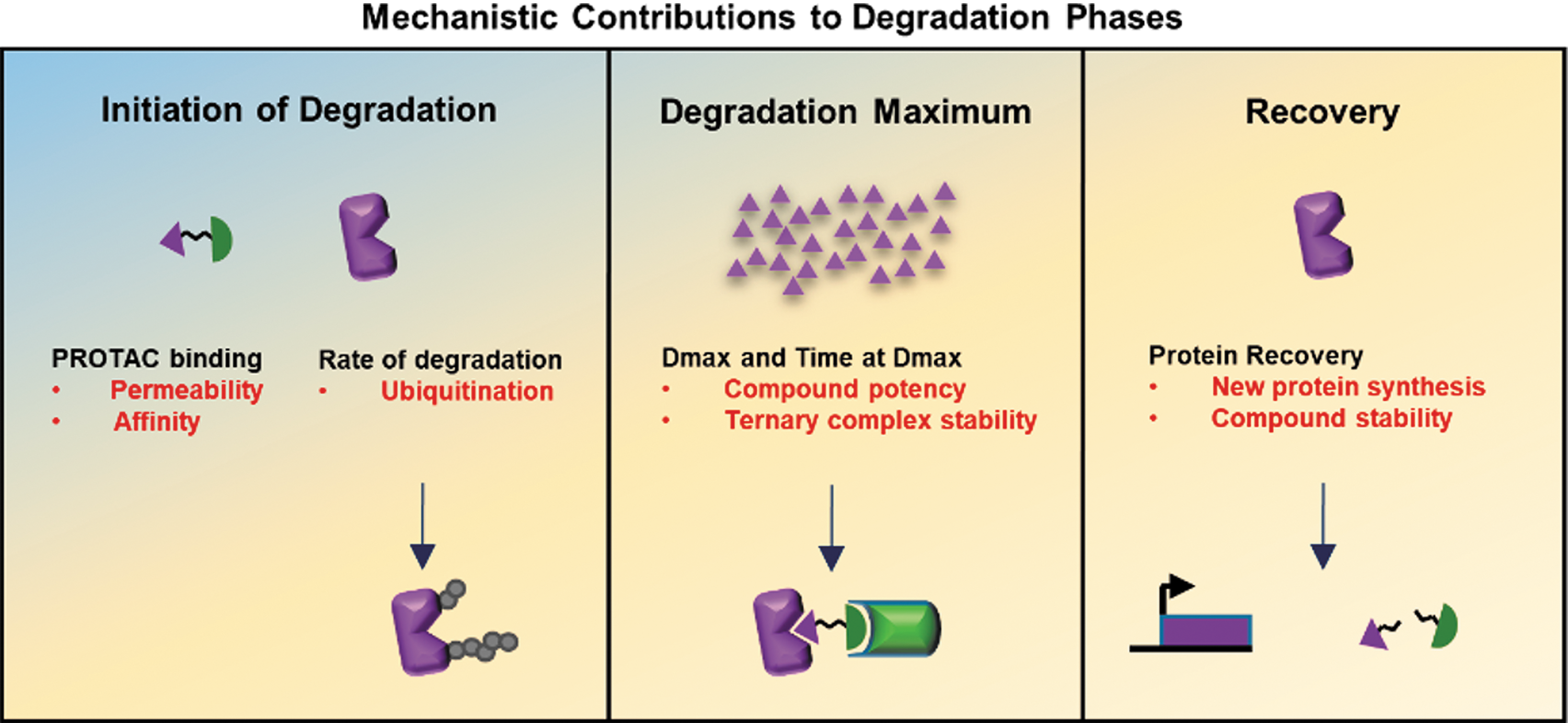
Commentary:Targeting proteins for degradation using bifunctional degrader compounds is an area of high interest in chemical biology and drug discovery research. This paper describes the use of Nanoluciferase (NanoLuc) to measure the three phases of protein degradation that occur when applying bifunctional degraders to a target protein (see
figure
). Described is the use of the HiBit split NanoLuc enzyme, which shows high affinity complementation via an 11 amino acid N-terminal fragment (HiBit) and the 18 kD enzyme fragment (LgBiT). To measure the initiation phase of degradation and the maximum degradation obtained (Dmax) as well as the recovery phase, CRISPR-Cas9 gene editing was used to introduce the HiBit peptide at the N-terminus of the protein of interest. The LgBiT is stably expressed in the cells using random integration. Upon addition of the degrader compound to live cells the kinetics can be followed using a luminometer (an Olympus LV200 microscope was employed). The kinetics show a degradation phase that fits to a single-component exponential decay model, which yields the degradation rate and Dmax values. The paper focusses on BET family members and employs the proteolysis targeting chimeras (PROTACs)—MZ1 and dBET1. Varying the concentration of the PROTAC demonstrates how different PROTACs act on different BET family members via the recruitment of E3 ligases. Addition of the BET inhibitor JQ1 then competes off the PROTAC and results in a measurable recovery phase. To measure ternary complex formation, NanoBRET technology is employed. In this case, the NanoBIT-tagged protein once complexed to a HaloTag E3 ligase (e.g., von Hippel Lindau or Cereblon) results in a BRET signal, providing measurement of target engagement kinetics. Therefore, this system provides important kinetic, potency, and stability information for PROTAC-mediated protein degradation. A key finding of the paper is that the MZ1/VHL recruitment system showed faster rates of degradation and slower but more stable ternary complex formation, with greater compound potency compared to the dBET1/CRBN system. Contributed by Doug Auld.
Model of degradation phases and contributing mechanisms Schematic showing the three phases of degradation: initiation of degradation, degradation maximum (Dmax), and recovery correlated to the key mechanistic processes listed in black for each phase. The target protein response to PROTACs at each phase is represented pictorially, showing first introduction of PROTAC and target protein, followed by loss of target protein, and then recovery. Listed in red are the parameters identified to regulate each phase with arrows depicting the processes which to monitor or optimize.
Scrolling Through Proteostasis
Schneider N, Gäbelein C, Wiener J, Georgiev T, Gobet N, Weber W, Meier M. Genetic code expansion method for temporal labeling of endogenously expressed proteins.ACS Chem Biol2018;13:3049–3053.
Abstract: We here present a method that combines genetic code expansion with CRISPR/Cas9 genome engineering to label endogenously expressed proteins with high spatiotemporal resolution. The method exploits the use of an orthogonal tRNA/tRNA synthetase pair in conjugation with noncanonical amino acids to create stop codon read through events. To demonstrate the functionality of the method, we pulse labeled endogenous β-actin and tumor protein p53 with a minimally invasive HA tag at their Ctermini. Targeting the protein label with a proximity ligation assay plus real time imaging facilitates seamless quantification of the protein synthesis rate and spatial localization at the single cell level. The presented approach does not interfere with any physiological control of cellular expression, nor did we observe any perturbation of endogenous protein functions.
Commentary:The mechanisms that control protein abundance involving both synthesis and degradation rates remain largely unknown for many proteins. This gap is in part due to the lack of technologies that can measure protein synthesis and degradation at either the population or single-cell level. This paper describes a method for labeling of endogenous proteins so that protein synthesis and degradation rates can be measured at the single-cell level. The method called SCROL (Stop-Codon-Read-thrOugh-Label) involves introducing an amber stop codon at the 3′ end of the gene locus for the protein of interest (POI). These engineered cells are then transfected with pyrrolysyl-tRNA synthetase (PylRS)/tRNA (CUA) pair, which incorporates non-coding amino acids (such as N(ɛ)-Boc-L-lysine) at amber codons, resulting in stop-codon read-through and expression of a tag to the POI (see
figure
). Protein synthesis and degradation rates can be measured by Western blots to the tag by adding N(ɛ)-Boc-L-lysine to the media in a series of pulse/chase experiments. The paper uses this to measure protein synthesis and degradation rates of β-actin and p53. To measure these rates at the single-cell level, an HA-tag was used to the POIs, and a proximity ligation assay (PLA) was carried out using anti-HA antibody followed by fixing and imaging of the cells.Enumerating the number of PLA dots within the cell population at each time point showed a bimodal distribution, which is consistent with protein synthesis rates being much faster during the cell cycle and the fact that the cells were not synchronized before the experiment. Live-cell imaging can also be used with SCROL, and this is demonstrated by tagging β-actin with Amber-mCherry. This method should be very useful in measuring proteostasis in cells. Contributed by Doug Auld.
Dynamic labeling of endogenously expressed proteins using SCROL. (A) Editing the DNA encoded protein label and Amber stop codon at the 3′ end of the targeted gene locus using the CRISPR/Cas9 system. (B) Transfection of the gene-edited cell with an orthogonal aminoacyl tRNA synthetase and cognate tRNA. (C) Temporal controlled labeling of the protein upon addition of an ncAA.