
Abstract
The retinoic acid-related orphan receptor gamma T (RORγt) plays an important role in Th17 cell proliferation and functionality. Thus, RORγt inverse agonists are thought to be potent therapeutic agents for Th17-mediated autoimmune diseases, such as rheumatoid arthritis, asthma, inflammatory bowel disease, and psoriasis. Although RORγt has constitutive activity, it is recognized that the receptor is physiologically regulated by various cholesterol derivatives. In this study, we sought to identify RORγt inverse agonists through a high-throughput screening campaign. To this end, we compared an apo-RORγt protein from Escherichia coli and a cholesterol-bound RORγt protein from insect cells. The IC50 of the known RORγt inverse agonist TO901317 was significantly lower for the apoprotein than for the cholesterol-bound RORγt. Through high-throughput screening using a fluorescence-based cholesterol binding assay with the apoprotein, we identified compound 1 as a novel cholesterol-competitive RORγt inverse agonist. Compound 1 inhibited the RORγt-TopFluor cholesterol interaction, coactivator recruitment, and transcriptional activity of RORγt. Cell-based reporter gene assay demonstrated that compound 1 showed higher potency by lipid depletion treatment. Collectively, our findings indicate that eliminating cholesterol from the RORγt protein is suitable for sensitive high-throughput screening to identify RORγt inverse agonists.
Introduction
The retinoic acid-related orphan receptor gamma T (RORγt) is a member of the nuclear receptor (NR) superfamily. 1 As an isoform of ROR gamma (RORγ), RORγt shares identical DNA binding domain (DBD) and ligand binding domain (LBD) sequences with RORγ. 2 However, expression of RORγt is restricted to several immune cell types, such as CD4+ Th17 cells, and is essential for the proliferation and functionality of Th17 cells. 3 Th17 cells produce interleukin-17 (IL-17) and induce tissue inflammation leading to the pathogenesis of various autoimmune diseases. 4 IL-17A expression is induced by RORγt, which directly binds to ROR response elements (ROREs) in the IL-17 promoter. 5 Hence, the differentiation of naive CD4+ T cells to Th17 cells is predominantly regulated by RORγt. 6,7
The liver X receptor agonist, TO901317 (Fig. 1A), has been identified as a potent RORγ inverse agonist 8 ; since then, many inverse agonists have been identified, with some reportedly inhibiting Th17 cell differentiation. Among these compounds, SR1001, ursolic acid, and digoxin have been found to suppress experimental autoimmune encephalomyelitis. 9 These recent reports strongly suggest that RORγt is a potential target for autoimmune diseases.

There has been considerable interest in endogenous ligands of RORs. X-ray crystal structure analysis has unexpectedly revealed that cholesterol was present in the ligand binding pocket (LBP) of ROR alpha (RORα) protein purified from insect cells. 10 Moreover, the crystal structure of the RORα protein, in complex with cholesterol sulfate, was solved following a ligand exchange experiment. 11 From the crystal structure of the RORβ protein expressed in Escherichia coli, stearic acids were identified as a fortuitous ligand. 12 In addition, the crystal structure of the RORγ LBD expressed in bacteria with a peptide motif of coactivator SRC1 and hydroxycholesterols has been solved. 13 These structures revealed that the RORs in complex possess active conformations that generate a hydrophobic coactivator binding surface. Recently, several oxysterols were found to be agonistic ligands of RORγ, 14 –18 suggesting that cholesterol biosynthetic intermediates may modulate the transcriptional activity of RORγ and RORγt.
To develop more sensitive assays for high-throughput screening (HTS), the selection of the appropriate recombinant protein is important. To date, limited reports exist about the relationship between the ROR protein and cholesterol. In 2002, Kallen et al. 10 reported that the RORα protein purified from Sf-9 cells was bound to cholesterol; however, the protein expressed in E. coli yielded only insoluble protein. They presumed that cholesterol, which was reportedly not synthesized in the bacteria, was needed for stabilizing the receptor. In addition, they reported that cholesterol could be exchanged with cholesterol sulfate in the RORα protein, and the exchange of bound cholesterol sulfate could not be reversed with the excess of other cholesterol derivatives. In contrast, Wang et al. 18 showed that the RORα protein expressed in E. coli could be reconstituted in a lipid-free environment and had the ability to bind coactivator peptides. These data suggest that cholesterol is not important for RORα protein stability or cofactor recruitment. In addition, these authors reported that the RORα protein treated with excess cholesterol sulfate blocked the binding of radiolabeled hydroxycholesterol to the receptor. Thus, whether the relationship between cholesterol and the recombinant RORα protein is applicable to RORγt remains unclear. In addition, there has been no evidence of the effect of endogenous cholesterol on RORγt inverse agonist activity for high-throughput screening.
In this study, we report that cholesterol within the LBP of the recombinant RORγt protein interferes with the potency of RORγt inverse agonist, and, by utilizing this result, a novel RORγt inverse agonist, compound 1, was identified through the high-throughput screening with TopFluor cholesterol binding assay. TAK-828F identified through further optimization of compound 1 has higher potency, and selectivity against NRs, including ROR isoforms. 19 –21 The compound also inhibited IL-17 secretion from mouse splenocytes and human primary cells, and inhibited Th17 cell differentiation from naive T cells and memory CD4+ T cells. 22 Furthermore, the compound showed significant efficacy in naive T cell transfer mouse colitis model.
Materials and Methods
Materials
TO901317 was purchased from Tocris Bioscience (Bristol, UK). Lovastatin and cholesterol were purchased from Wako Pure Chemical Industries (Osaka, Japan). Cholesterol sulfate was purchased from Sigma-Aldrich Corporation (St. Louis, MO).
Establishment of Stable Cell Lines for Reporter Gene Assay
The reporter-gene construct, pGL4.28-hIL17-ROREx3-luc, was constructed as described by Ichiyama et al. 23 Briefly, the construct was created by inserting an oligonucleotide, comprising three copies of the RORE in the human IL17 promoter, into the pGL4.28 [luc2CP/minP/Hygro] reporter vector (Promega Corporation, Fitchburg, WI). The full-length complementary DNA for human RORγt was subcloned into the mammalian expression vector pMCMV-neo, which contained the cytomegalovirus promoter, to generate pMCMVneo-RORγt. To generate stable cell lines, pGL4.28/hIL17-ROREx3-luc and pMCMVneo-RORγt were transfected into the Jurkat Tet-On cell line (Takara Bio, Inc., Kusatsu, Japan) using Gene Pulser (Bio-Rad Laboratories, Inc., Hercules, CA), and stable cell lines were chosen based on luciferase activity.
Protein Expression and Purification
The DNA fragment of human RORγt LBD (Uniprot: P51449, aa. 261–518), amplified by PCR, was cloned into the pFastBacHTb vector (Thermo Fisher Scientific, Waltham, MA). Recombinant baculovirus was generated using the Bac-to-Bac baculovirus expression system (Thermo Fisher Scientific). Protein expressed in Sf9 insect cells was harvested 48 hours postinfection. Cell pellets were homogenized in a lysis buffer [50 mM Tris-HCl, pH 7.9, 200 mM NaCl, 5% glycerol, 0.25 mM Tri(2-carboxyethyl) phosphine (TCEP), and Complete™ protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland)]. Cell lysates were clarified by high-speed centrifugation at 37,000 g for 45 min and applied to affinity chromatography with ProBond Ni-chelating resin (Thermo Fisher Scientific). After extensive washing, the His-tagged protein was eluted by 50 mM Tris-HCl, pH 7.9, 150 mM NaCl, 200 mM imidazole, and 1 mM TCEP. The protein was then buffer exchanged into 25 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM dithiothreitol (DTT), and 1 mM TCEP before loading onto a MonoQ column (GE Healthcare, Chicago, IL). The flow-through fraction containing the LBD was collected. Protein concentration was measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific). The purity of the protein sample was verified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and liquid chromatography–mass spectrometry (LC-MS) (Micromass LCT™ Workstation; Waters Corporation, Milford, MA).
A truncated human RORγt LBD (aa. 261–508) with an N-terminal 6xHis tag, followed by a TEV cleavage site, was cloned into a modified pET vector and expressed in E. coli BL21 (DE3) cells. The bacterial culture was grown in LB media at 37°C. When the culture reached an OD600 of 0.8, the cells were induced with 0.8 mM IPTG for an additional 18-h growth at 16°C before harvesting. The cell pellet was resuspended and sonicated in lysis buffer [25 mM Tris-HCl, pH 7.6, 1 M NaCl, 10 mM imidazole, 0.5 mM TCEP, 20 U/mL benzonase, 1 mg/mL lysozyme, and Complete™ protease inhibitor cocktail (Roche Diagnostics)]. The cell lysate was clarified by centrifugation (37,000 g, 45 min, and 4°C) and applied to a 5 mL HiTrap Talon column (GE Healthcare). After an extensive wash, the His-tagged protein was eluted from the column with buffer containing 300 mM imidazole in 25 mM Tris-HCl, pH7.6, 1 M NaCl, 2 mM benzamidine, and 0.5 mM TCEP. The protein sample was buffer exchanged into 25 mM Tris-HCl, 200 mM NaCl, 2 mM benzamidine, 2 mM DTT, and 5% glycerol. It was then concentrated and loaded onto a size exclusion chromatography column (Hiload 16/60 Superdex 200; GE Healthcare). Peak fractions containing the truncated LBD protein were pooled and concentrated to 1 mg/mL. The purity of the final protein sample was verified by SDS-PAGE and LC-MS. The concentration of RORγt proteins were measured by Bradford Protein assay (Bio-Rad).
Quantification of Cholesterol by Mass Spectrometry
Protein samples were dissolved in 0.5% SDS solutions to a concentration of 500 nM, and were heated at 95°C for 5 min to denature the sample completely. For this experiment, a Shimadzu HPLC system (Shimadzu Corp., Kyoto, Japan) and API 5000 triple quadrupole mass spectrometer (AB SCIEX, Framingham, MA) were used to quantify the concentration of cholesterol. The analysis of cholesterol was performed using a Unison UK-C8 30 × 3 mm ID column, 3 μm (Imtakt, Kyoto, Japan). The HPLC mobile phase A contained 10 mM ammonium acetate in water, while mobile phase B contained 10 mM ammonium acetate in acetonitrile. The chromatographic separation of cholesterol was conducted using a constant ratio of 93% of mobile phase B in mobile phase A at a flow rate of 0.8 mL/min. According to the previous report for determination of 7α-OH cholesterol, the [M+H]+ ions are instable at high temperature in the ionization step, and its daughter ions losing a water molecule are more stable. 24 So, the concentration of cholesterol was determined with selected monitoring methods and precursor ion scanning of m/z 369.3 (in a form of [M+H-H2O]+) in positive ion mode specific for cholesterol.
Reporter Gene Assay
The principle of the assay is outlined in Figure 1B. Stable Jurkat cells were maintained in RPMI 1640 medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin G, and 100 μg/mL streptomycin sulfate (Thermo Fisher Scientific). In experiments, cells were plated in 384-well white tissue culture plates (3570; CORNING Incorporated, Corning, NY) with 1 μg/mL of doxycycline (Takara Bio) at a density of 20,000 cells per well. For cholesterol-depletion treatment, cells were maintained on RPMI 1640 containing 10% lipid-reduced FBS (GE Healthcare) and 10 μM lovastatin. Diminishing intracellular cholesterol was confirmed using a cholesterol fluorometric assay kit (Cayman, Ann Arbor, MI) (data not shown). After culturing for 3 h, cells were treated with dimethyl sulfoxide (DMSO) or test compounds and incubated at 37°C for 20 h. Luciferase activity was measured by Envision (PerkinElmer, Waltham, MA) using the Bright-Glo Luciferase Assay System (Promega). Raw data were analyzed using Prism 5 (GraphPad Software, San Diego, CA), and a three-parameter logistic fit equation [Eq. (1)] was used to determine IC50.
“X” is the log of compound concentration and “Y” is response.
For normalization of data in Table 3, the cells treated with 1 mg/mL doxycycline wells were used as 100% controls (top) and the cells without doxycycline were used as 0% controls (bottom). IC50 values were calculated by Equation (1) using Prism 5.
TopFluor Cholesterol/RORγt LBD (Time-Resolved Fluorescence Resonance Energy Transfer) Binding Assay Screening
The principle of the assay is outlined in Figure 1C, and the detailed protocol is described in Table 1. In 384-well black low-volume plates (Greiner Bio-One, Kremsmünster, Austria), 3 μL per well of the library compounds diluted with an assay buffer [20 mM Tris-HCl (pH7.5), 100 mM NaCl, 1 mM DTT, 0.1% bovine serum albumin (BSA)] was added to a concentration of 1 μM. Final DMSO concentration was 1%. The plates were then incubated at room temperature for 20 min after the addition of 3 μL/well of RORγt protein expressed from E. coli at a final concentration of 60 nM. TopFluor cholesterol (3 μM; Avanti Polar Lipids, Alabaster, AL) and anti-His Terbium (Tb) (2 nM; Thermo Fisher Scientific) were then added to each well at 3 μL/well. An HT station 1200 (MSTechnos, Tokyo, Japan) was used to transfer the compounds, and a Multidrop Combi (Thermo Fisher Scientific) was used to transfer the RORγt protein, TopFluor cholesterol, and anti-His Tb. The plates were incubated at room temperature for 1 h and measured in Envision. To eliminate false positive compounds, a Fluorescein-His Tag (2 nM; Thermo Fisher Scientific) was used instead of the RORγt protein and TopFluor cholesterol. The Tb donor was excited at 320 nm, its emission was monitored at 486 nm, and the acceptor emission was monitored at 520 nm. The results were expressed as a ratio of Em520/Em486. Raw data were analyzed using Prism 5, and a three-parameter logistic fit equation [Eq. (1)] was used to determine IC50. For normalization of the data in Table 3 and Figure 3C, the wells containing DMSO were defined as 0% inhibition control (bottom) and the wells containing 10 μM of TO901317 were defined as 100% inhibition control (top). IC50 values were calculated in Equation (1) using Prism 5.
TopFluor Cholesterol/RORγt Ligand Binding Domain (Time-Resolved Fluorescence Resonance Energy Transfer) Binding Assay Screening Protocol Table
Columns 1–20, library compounds at 1 μM; columns 21–22, 1% DMSO; columns 23–24, 1% DMSO plus 10 μM of TO901317.
Spin plates for 10 s at 1,000 rpm, cover the plate before incubation.
Read the plate in an Envision (PerkinElmer).
DMSO, dimethyl sulfoxide; RORγt, retinoic acid-related orphan receptor gamma T; Tb, Terbium; TR-FRET, time-resolved fluorescence resonance energy transfer.
Cell-Free Cofactor Peptide Recruitment Assay
The principle of the assay is outlined in Figure 1D. The binding of a cofactor peptide motif to the purified RORγt protein was determined using the AlphaScreen Histidine (Nickel Chelate) Detection Kit (PerkinElmer). The experiments were conducted with 50 nM RORγt LBD and 10 nM of biotinylated SRC1-2 peptide (CPSSHSSLTERHKILHRLLQEGSPS, Scrum, Tokyo, Japan) in the presence of 10 μg/mL donor and acceptor beads in buffer containing 50 mM Tris-HCl (pH7.4), 50 mM KCl, 1 mM DTT, and 0.1% BSA. Assay plates were incubated for 3 h at room temperature and measured by Envision. Raw data were analyzed using Prism 5, and a three-parameter logistic fit equation [Eq. (1)] was used to determine IC50. For normalization of the data in Table 3, the wells containing DMSO were defined as 100% control (top) and the wells without the RORγt protein were defined as 0% control (bottom). IC50 values were calculated in Equation (1) using Prism 5.
Thermal Shift Assay
In 96-well V-bottom polypropylene microplate (3363; CORNING), 20 μL/well of the test compounds dissolved in an assay buffer [20 mM Tris-HCl (pH7.5), 100 mM NaCl, 1 mM DTT] was added with 20 μL per well of the RORγt protein expressed from E. coli (3 μM) and 20 μL/well of SYPRO Orange (500-fold diluted; Thermo Fisher Scientific). Then, 8 μL per well of the reaction was transferred to a 384-well PCR plate in quadruplicate. After centrifuging, the plates were heated at a ramp rate of 1°C/min and the fluorescence was monitored by ABI 7900 (Thermo Fisher Scientific). Data were analyzed by Prism 5 (GraphPad Software), and Boltzman sigmoidal fitting was used to determine melting temperature (Tm ).
Results
Selection of the Human RORγt Protein for HTS
To identify lead compounds of NRs, cell-based reporter gene assay, cell-free ligand binding assay, and cell-free cofactor peptide recruitment assay are general assay methods. 25 Cell-based assays can measure transcriptional effect of a compound; however, they detect a lot of false positive compounds such as toxic compounds or indirect regulators like kinase inhibitors. In contrast, cell-free assays are, although they do not exactly reflect the target in the cellular context, simple and more likely to identify direct binding modulator. So, we developed a cell-free assay system for RORγt inverse agonist screening. To select the protein suitable for primary screening, we produced two types of human RORγt protein, expressed in E. coli [RORγt (E. coli)] and in insect cells [RORγt (Ins)], and compared the presence of cholesterol in the LBD, ligand binding activity, cofactor recruitment activity, and tool compound effect.
At first, we performed mass spectrometry analysis for measuring the cholesterol content of the proteins. As a result, more than 60% of the RORγt (Ins) protein was found to contain cholesterol. In contrast, the RORγt (E. coli) protein did not contain any cholesterol, although there was a possibility that the protein would contain cholesterols incorporated from the medium during the cultivation of E. coli cells (Table 2). Furthermore, we compared the activity of these proteins using the TopFluor cholesterol competition assay (Fig. 1C) and cofactor recruitment assay (Fig. 1D). Generally, an agonist binding to NR causes conformational change resulting in an interaction of coactivator for transcription of target genes. RORγt is constitutive active and can interact with a coactivator such as SRC1 without ligands. 13 So, we used cell-free cofactor recruitment assay to determine whether the RORγt protein is in an active or inactive state by detecting interaction of SRC1-2 peptide with the RORγt LBD. As shown in Figure 2A, both RORγt (E. coli) and RORγt (Ins) proteins were effective in recruiting the SRC1-2 peptide motif. Exogenous cholesterol weakly promoted to the SRC1 peptide recruitment by RORγt (E. coli) and the EC50 was about 2.5 μM, while its activation was not observed in the reporter gene assay (data not shown). This observation implies that cholesterol is a very weak agonist, not affecting the transcriptional activity of RORγt.

Cholesterol binding activity and SRC1–2 peptide recruitment activity of the three RORγt proteins. RORγt (Ins), RORγt (Escherichia coli), and cholesterol sulfate-saturated RORγt (E. coli) were analyzed by cofactor peptide recruitment assay and TopFluor cholesterol binding assay.
Cholesterol Concentration in the RORγt Protein Expressed in Insect Cells or Escherichia coli as Measured by Mass Spectrometry
To investigate cholesterol binding activity of these proteins, we developed a time-resolved fluorescence resonance energy transfer (TR-FRET) binding assay, which detects TopFluor cholesterol binding to the RORγt LBD (Fig. 1C). As shown in Figure 2B, the RORγt (E. coli) protein could specifically bind TopFluor cholesterol compared to no protein control and the apparent Kd was calculated as 6.3 μM, while the binding of the RORγt (Ins) was only detected at <10% of the RORγt (E. coli) binding capability. Exogenous cholesterol can displace the TopFluor cholesterol in RORγt (E. coli) LBD and its IC50 was about 7.2 μM.
We then examined whether differences in host cells affect the potency of the inverse agonist activity using TO901317, which is a known RORγt inverse agonist. As shown in Figure 2C, the SRC1-2 recruitment assay revealed that TO901317 was over 200 times more potent on RORγt (E. coli) (IC50 = 23 nM) than on RORγt (Ins) (IC50 = 6,700 nM). Crystal structure of the LBD of RORγ with TO901317 showed that binding site of this compound is same as cholesterol binding site. 26 To corroborate this structural information, ligand competitive assay for TO901317 was conducted in TopFluor cholesterol binding assay. As expected, the IC50 value of TO901317 was right-ward shifted depending on increasing TopFluor cholesterol concentration (Fig. 2D). It is difficult to increase TopFluor cholesterol concentration more than 10 μM (about twofold higher than Kd ) because of its solubility. These data demonstrate that TO901317 inhibited TopFluor cholesterol binding in a competitive manner.
To determine whether the reason was due to the difference in expression system or the presence of cholesterol, we tested the activity of RORγt (E. coli) incubated with excess cholesterol sulfate and applied to gel filtration to remove free cholesterol sulfate. Wang et al. 18 used cholesterol sulfate for examining the effect of exogenous sterol to the RORα LBD. So, cholesterol sulfate was used as a surrogate ligand because it had more solubility and higher affinity for RORγt (IC50 = 46 nM data not shown) than cholesterol, and it is thought to be a nonfunctional ligand like cholesterol in cofactor recruit assay and reporter gene assay according to a previous report. 18 Cholesterol sulfate-saturated RORγt (E. coli), as well as RORγt (Ins), blocked the binding of the TopFluor cholesterol (Fig. 2B) and inhibition by TO901317 in the cofactor recruitment assay (IC50 > 10,000 nM, Fig. 2C), indicating that bound cholesterol sulfate is hardly exchanged from the RORγt protein. From these results, we selected the apo-RORγt (E. coli) protein for primary screening because it was expected that using the apoprotein would identify a greater variety of hit clusters of RORγt inverse agonists.
High-Throughput Screening
Among two established cell-free assays, we chose the TopFluor cholesterol competition assay for primary screening to identify not only inverse agonists but also antagonists, which cancel the agonistic activity of cholesterol derivatives. TopFluor cholesterol concentration was set as 3 μM according to the apparent Kd from titration data (Fig. 2B) and assay robustness. An assay validation such as duplicate (N2) correlation and batch reproducibility was conducted, and test concentration was set as 1 μM based on a hit rate of prescreening (data not shown).
A high-throughput screening was conducted on the 860,000 compounds of the Takeda compound library at 1 μM as outlined in Figure 3A. From a scatterplot of 1% DMSO controls and TO901317 controls in a single representative data from the primary screening (Fig. 3B), Z’-factor was calculated as 0.86. The average Z’-factor of all plates tested was 0.80 ± 0.05 [average ± standard deviation (SD)] with an average plate S/B ratio of 10.80 ± 1.23. The primary screening data in histogram format (Fig. 3C) showed a normal distribution curve with an average inhibition value of 10.9% and an SD of 17.2%. Since T0901317 showed over 15-fold less activity in cell-based assay [IC50 (reporter gene assay) = 1,500 nM] (Table 3) than cell-free assay [IC50 (TopFluor cholesterol binding assay) = 91 nM, IC50 (cofactor recruitment assay) = 23 nM] (Table 3), we set a cutoff criteria of 70% inhibition at 1 μM to identify cellular active compounds directly from HTS campaign. The remaining compounds above 70% at 1 μM from primary screening were tested in a counter assay using a Fluorescein-His tag to eliminate false positive compounds due to the inhibition of the TR-FRET signal. In total, 229 compounds passed the counter assay, and from these, 68 compounds showed >70% inhibition against the apo-RORγt-SRC1 peptide interaction at 1 μM. Following RORγt-RORE reporter gene assay, 23 clusters (41 compounds) were identified as hit compounds.

Primary screening results.
IC50 Values of Compound 1 and T0901317 in RORγt Reporter Gene Assay, TopFluor Cholesterol Binding Assay, and Cofactor Recruitment Assay Using Apo-RORγt (Escherichia coli)
For reporter gene assay, compounds were tested in duplicate in medium containing lipid-reduced FBS with 10 μM lovastatin. IC50 values were calculated as described in the Materials and Methods section using normalized data from a single experiment performed in duplicate.
CI, confidence interval; FBS, fetal bovine serum.
Of these hit compounds, compound 1 (Fig. 3D) was selected as a lead compound because the compound had 15-fold greater selectivity against RORα and RORβ, 19 has good physical properties, including water solubility and cLogP of 2.8, and did not have cytotoxity and CYP inhibition. Table 3 summarizes the IC50 values of compound 1 and TO901317 for RORγt from three functional assays, demonstrating that compound 1, similar to TO901317, was effective in the TopFluor cholesterol binding assay (IC50 = 200 nM), the SRC1-2 recruitment assay (IC50 = 91 nM), and the RORγt-RORE reporter gene assay (IC50 = 1,600 nM), although the potency of compound 1 was slightly weaker than TO901317.
To confirm the direct interaction between compound 1 and RORγt, we performed the thermal shift assay (TSA). TSA is a common method to confirm direct binding of small molecule to the target protein, including RORγ. 14,27 TSA monitors the thermal stability of the target protein with fluorescent dye such as SYPRO Orange, which emits fluorescence when hydrophobic residues of the protein are exposed to the dye. A ligand-induced conformational stabilization of the target protein can be judged by comparing Tm with or without ligands. As shown in Figure 4A and B, Tm value of TO901317 (51.6°C at 30 μM) and compound 1 (51.7°C at 30 μM) was higher compared with the apo-form of RORγt (E. coli) (Tm = 45.3°C). Tm shift by compound 1 at 3 μM (2.4°C) was lower than by TO901317 (3.6°C), consistent with their order of IC50 in cell-free assay. So direct binding of compound 1 was confirmed in TSA.

Direct binding of TO901317 and compound 1 by thermal shift assay. Representative melting curves of RORγt (E. coli) with 0, 3, 10, or 30 μM TO901317
The Effect of Cholesterol on the Potency of Compound 1 in Cell-Free and Cell-Based Assay
To determine the effect of cholesterol on the inhibition of compound 1, cell-free SRC1-2 recruitment assay and cell-based reporter gene assay were conducted. In the SRC1-2 recruitment assay, similar to TO901317, the IC50 of compound 1 was over 100-times lower for apo-RORγt (E. coli) than for RORγt (Ins) and cholesterol sulfate-bound RORγt (E. coli), as shown in Figure 5A. These results revealed that compound 1 could not have been identified if RORγt (Ins) was used for primary screening. Figure 5B showed that compound 1 inhibited TopFluor cholesterol binding of RORγt protein in a competitive manner, same as TO901317, and further structural analysis of compound 1 derivative revealed that this chemotype binds to cholesterol binding site. 19

The effect of cholesterol on the potency of compound 1 in SRC1-2 peptide recruitment assay and reporter gene assay.
To confirm the effect of cholesterol for RORγt transcriptional activity, we used RORE reporter gene assay (Fig. 1B), detecting inhibition of RORγt's constitutive active transcriptional activity by inverse agonist. Compound 1 was then tested with normal medium, cholesterol-depleted medium (lipid-reduced FBS and 10 μM of lovastatin), or cholesterol-depleted medium containing cholesterol sulfate. The result showed that cholesterol depletion and cholesterol sulfate treatment had almost no effect against RORγt transcriptional activity, suggesting that cholesterol and cholesterol sulfate are nonfunctional ligands (Fig. 5C), while, as shown in Figure 5D, the IC50 value of compound 1 was 3.1 μM with cholesterol depletion treatment, which was lower than the IC50 value of 14 μM without cholesterol depletion. Furthermore, increasing amounts of cholesterol sulfate in the cholesterol-depleted medium induced a rightward IC50 shift, although the transcriptional activity without compound 1 did not increase with cholesterol sulfate. These results strongly suggest that the IC50 shift in the inverse agonist activity by lovastatin arose from the intracellular cholesterol concentration, not an indirect effect of lovastatin.
From these results, in cell-free-assay, we concluded the mechanism of action of RORγt inverse agonists (Fig. 6). TO901317 and compound 1 bind to LBP of apo-RORγt protein to induce the inactive form of the protein. That results in inhibiting the recruitment of SRC1 peptide to the protein. In contrast, cholesterol-bound RORγt protein is hardly exchanged by these RORγt inverse agonists and sustains the active form of the protein and the recruitment of SRC1 peptide. Furthermore, in the cell-based assay, the intracellular cholesterol level affected the potency of the RORγt inverse agonist. Therefore, it is noteworthy that reducing cholesterol levels in both cell-free and cell-based assay is necessary for the sensitive screening of RORγt inverse agonists.
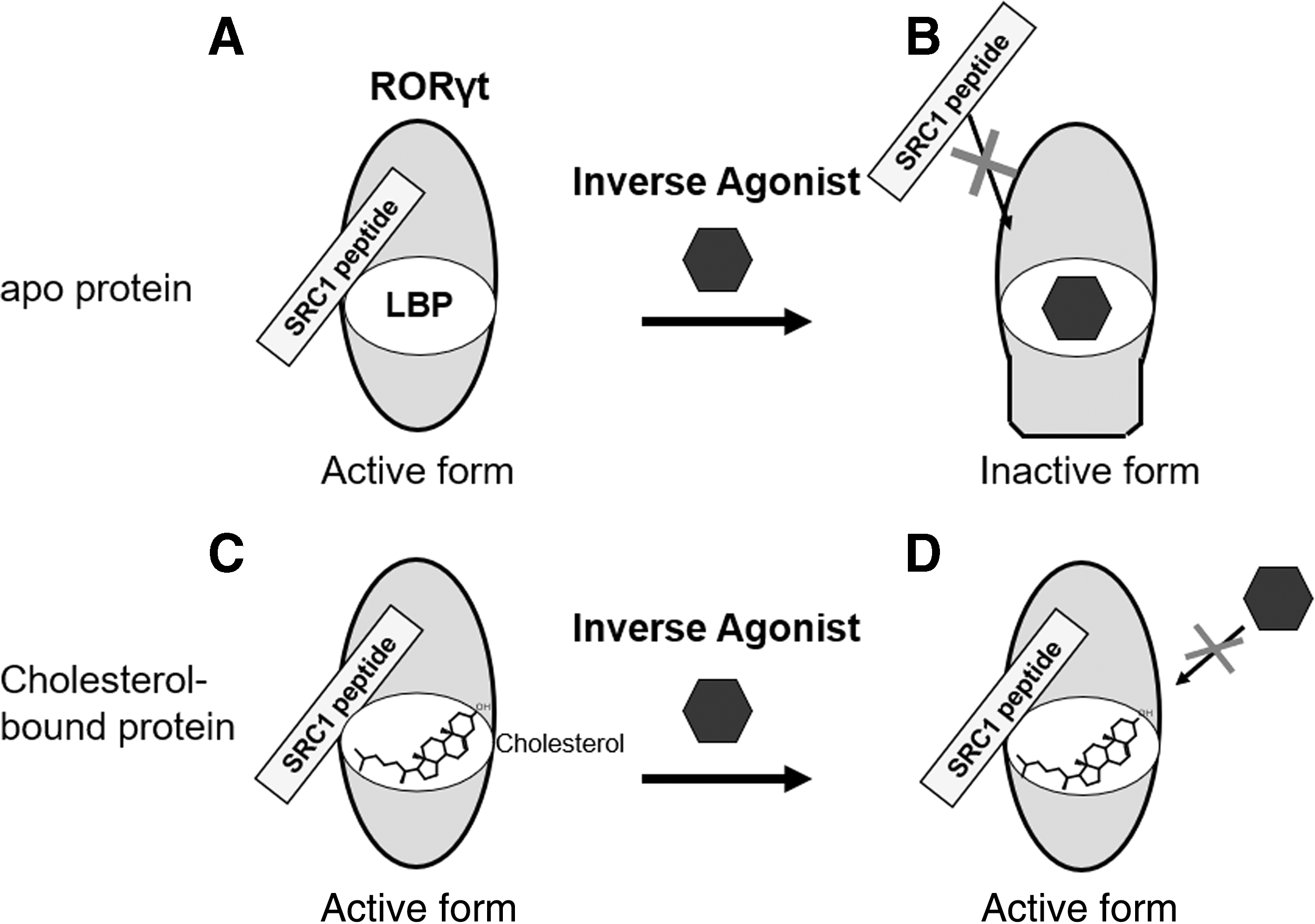
The effect of cholesterol against the recombinant RORγt protein and RORγt inverse agonist. Cholesterol-free apoprotein has constitutive activity and can interact with SRC1 peptide
Discussion
We have shown using cell-free assays that TO901317 was more potent on the RORγt (E. coli) protein than on the RORγt (Ins) protein due to the difference in their cholesterol content. In addition, compound 1 was identified as a RORγt inverse agonist through TopFluor cholesterol binding screening with the apo-RORγt protein. In the cell-based reporter gene assay, reducing intracellular cholesterol levels lowered the IC50 of compound 1. Although the apo-RORγt protein expressed in E. coli was already used in in vitro functional assays, and cholesterol depletion treatment has been used in cell-based assays by different authors, 14,18 the results obtained in this study show that the effect of cholesterol on the potency of RORγt inverse agonists is large. Thus, excluding cholesterol is important for sensitive assays to conduct high-throughput screening. In fact, high-throughput screening using SRC1-2 recruitment assay with a small-scale library with the RORγt (Ins) protein yielded no positive compounds (data not shown).
Although our data using LC/MS showed that ∼60% of RORγt (Ins) is bound to cholesterol, TopFluor cholesterol could bind <10% of the RORγt (Ins). This discrepancy is thought to be attributed to the presence of other cholesterol derivatives because we detected the specific peak for cholesterol. This observation is similar to the report by Bitsch et al. 28 showing that 77% of cholesterol and 18% of 7-dehydrocholesterol were bound with RORα LBD protein expressed in Sf9 cells. Like RORγt (Ins) protein, cholesterol sulfate-bound RORγt (E. coli) blocked the binding of fluorescence-labeled cholesterol and the inverse agonists, which is consistent with the previous report by Wang et al. 18 illustrating that cholesterol sulfate-saturated RORα-LBD inhibited the binding of 3 H-25OHC to the receptor.
We showed that the presence of cholesterol within the RORγt (Ins) interfered with binding of the RORγt inverse agonist; however, IC50 of cholesterol in TR-FRET binding assay was higher (about 7.2 μM) than we expected. According to this result, we assumed that cholesterol interacts with RORγt in a time-dependent manner. To prove the possibility of slow binding kinetics of cholesterol against RORγt, we tried to perform SPR experiment by using Biacore biosensor, but it did not work because of sticky profile of cholesterol. Further experiments are required to investigate the binding mode of cholesterol against RORγt.
As for the cell-free assay, we showed that compound 1 and TO901317 could not exchange the endogenous cholesterol bound to the RORγt protein (IC50 of compound 1 was >10 μM), while in the cell-based RORγt reporter gene assay, they inhibited the RORγt transcriptional activity (IC50 of compound 1 was 14 μM) in normal FBS medium, in which intracellular cholesterol is abundant, and IC50 ratio of normal condition versus cholesterol depletion treatment in the reporter gene assay was lower than cell-free assay. In addition, various inverse agonist compounds, including ursolic acid, and digoxin are effective in in vivo assay, although endogenous cholesterol is present, 29 suggesting that LBP of RORγt in mammalian cells is exchangeable by inverse agonists. We speculated one reason that the intracellular turnover of RORγt is faster compared with other cellular proteins. In fact, our SILAC analysis showed that the half-life of RORγt was ∼6.2 h (data not shown). On the time scale of the cell-based assay, RORγt inverse agonists may bind to RORγt before cholesterol binds the protein generated in the cell body. Another possible reason is that cholesterol derivatives bound to RORγt in mammalian cells can be exchanged by the inverse agonists because of weaker affinity. This observation in which ligand bound with purified protein acts as a potency tuner is supported by the previous reports with hepatocyte nuclear factor 4 (HNF4) family, which show high constitutive activity. Crystallographic analysis of HNF4α and HNF4γ LBD protein identified a mixture of fatty acids 30,31 and the endogenous fatty acids bound with the recombinant protein did not readily exchange with radiolabeled palmitic acid. On the other hand, affinity isolation/mass spectrometry showed that endogenously expressed HNF4α in mammalian cells was occupied by linoleic acid. This ligand was silent ligand and linoleic acid-bound HNF4α protein was exchangeable by another ligand in mammalian cells. 32 These reports suggest that composition of ligands bound with the NR protein is different in host cells, and further investigation of the factual form of the RORγt LBD in mammalian cells should be conducted.
In summary, we identified compound 1 as a cholesterol-competitive RORγt inverse agonist and showed that the inverse agonist effect of compound 1 depends on whether cholesterol is saturated in the receptor or not. Further compound optimization study of compound 1 has been reported by Shirai et al. 19 This study emphasizes the importance of characterizing the recombinant protein and the optimization of cell-based assay conditions for high-throughput screening, and should impact the investigation of other orphan NRs for obtaining new lead compounds.
Footnotes
Acknowledgments
The authors thank Shoichi Okubo and Makiko Kawamoto for preparation of plasmids and stable cell line construction. They also thank Akiko Oki for SILAC analysis. The authors also thank Yuji Shimizu for helpful discussion and Junji Matsui and Naoki Tarui for their support in this study. This study was conducted with financial support from Takeda Pharmaceutical Company, Ltd. and SCOHIA PHARMA, Inc.
Disclosure Statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.