
Abstract
In response to a variety of insults the unfolded protein response (UPR) is a major cell program quickly engaged to promote either cell survival or if stress levels cannot be relieved, apoptosis. UPR relies on three major pathways, named from the endoplasmic reticulum (ER) resident proteins IRE1α, PERK, and ATF6 that mediate response. Current tools to measure the activation of these ER stress response pathways in mammalian cells are cumbersome and not compatible with high-throughput imaging. In this study, we present IRE1α and PERK sensors with improved sensitivity, based on the canonical events of xbp1 splicing and ATF4 translation at ORF3. These sensors can be integrated into host cell genomes through lentiviral transduction, opening the way for use in a wide array of immortalized or primary mammalian cells. We demonstrate that high-throughput single-cell analysis offers unprecedented kinetic details compared with endpoint measurement of IRE1α and PERK activity. Finally, we point out the limitations of dye-based nuclear segmentation for live cell imaging applications, as we show that these dyes induce UPR and can strongly affect both the kinetic and dynamic responses of IRE1α and PERK pathways.
Introduction
Response and adaptation to environmental cues is one of the key features of any biological system. At the cellular level, mammalian cells adjust their metabolic and biosynthesis output depending on the resources available. The endoplasmic reticulum (ER), a major hub for protein biosynthesis within the cell, is in charge of the “quality control” of synthesized proteins and plays an essential role by ensuring their correct three-dimensional folding. 1,2 When this machinery is overloaded, failsafe mechanisms are triggered to alleviate ER stress, namely the unfolded protein response (UPR). UPR relies on three “sensing” proteins at the ER membrane, IRE1α, PERK, and ATF6, which integrate stress cues to make a decision between activating transcriptional programs to repair damage and committing to death by apoptosis. 3 Upon ER stress, the chaperone BiP/Grp78 is displaced from its binding site on IRE1α, PERK, and ATF6 at the ER lumen, releasing these proteins to execute their downstream signaling pathways. Once triggered, IRE1α RNase has been shown to remove a 26 bp intron from xbp1 mRNA that leads to the translation of a functional XBP1 transcription factor, activating mostly prosurvival downstream targets. 4 PERK activation leads to its dimerization and phosphorylation of eIF2α, a translation initiation factor that can switch the initiation codon of the ATF4 protein from the open reading frame (ORF)1 and 2 to ORF3, as well as decreasing the overall amount of protein synthesis. 4 ATF4 translation at ORF3 leads to a functional ATF4 protein, activating the transcription of prosurvival targets, but also proapoptotic proteins from the Bcl-2 family, such as Bim, Puma, or Noxa. 4,5
The UPR plays a critical part not only in many pathologies, such as cancer, kidney diseases, or neurodegenerative disorders, but also ischemia/reperfusion injuries such as strokes. 6 –9 As a result, the regulatory pathways have received considerable attention and recently, small-molecule inhibitors have been designed to block IRE1α RNase activity or PERK kinase activity for therapeutic intervention in human pathologies. 10,11 To date, these drugs have serious side effects, thus, there is a need for improved ATF4 and XBP1 sensors for the identification of novel selective drugs for the pharmacological modulation of these pathways. 12,13
However, a consensus method to detect the dynamic response of the ER stress pathways compatible with high-throughput screening applications is still lacking. Canonical methods to detect ER stress (i) primarily consisted of the use of endpoint assays, that is, western blot to detect the expression of ATF4 targets, such as C/EBP homologous protein (CHOP) or quantitative reverse transcription polymerase chain reaction to detect the expression spliced isoform of xbp1 mRNA. 14 Other reported methods (ii) used indirect bioluminescence transcription reporter assays composed of ATF4, XBP1, or ATF6 response elements engineered to regulate the expression of a luciferase gene. 15 We decided to focus on the improvement of a third category of UPR activation sensors (iii) that have been reported to exploit xbp1 splicing-induced frameshift 16 or ATF4 ORF switch 17,18 to express a fluorescent protein in-frame. Thanks to the recent discovery of very bright fluorescent proteins in the green 19 and red spectrum 20 ; it is now possible to build a system to directly detect the early UPR response dynamics that is so important to determine cell fate.
In the present study, we report the use and validation of a new-generation dual fluorescent reporter system for ATF4 translation and xbp1 splicing for time-lapse imaging. We show that this assay can be multiplexed using different wavelengths and performed using commercially available high-content imagers. In addition, we demonstrate that the use of nuclear dyes for nuclear segmentation in time-lapse experiments should be strongly discouraged, as they drastically modify the response of the IRE1 and PERK signaling pathway in the presence or absence of stress induction.
Materials and Methods
Reagents
Thapsigargin was purchased from Enzo Life Science (BML-PE180). Hoechst 33342 was purchased from Cell Signaling technology (#4082). DRAQ5™ was purchased from Thermo Fisher Scientific (62252). Dimethyl sulfoxide (DMSO) was purchased from Fisher Scientific (BP231).
Western Blot
Western blot was performed as previously published 21 with the following primary antibodies: Vinculin (SC-55465; Santa Cruz Biotechnology), human influenza hemagglutinin (HA) (MMS-101P; Covance), and CHOP (#2895; Cell Signaling Technology).
DNA Constructs
Synthetic sequences of ATF4 mScarlet nuclear localization sequence (NLS) (PERK sensor) and XBP1 mNeonGreen NLS (IRE1 sensor) were ordered from Integrated DNA Technologies as linear double-stranded DNA and cloned into pLVx vector using EcoRI and XbaI restriction sites, or into pLHCx vector using HindIII and ClaI restriction sites.
Cell Lines and Cell Culture Conditions
Human cervical adenocarcinoma cells (HeLa; ATCC), human colorectal carcinoma cells (HCT116; ATCC), and human embryonic kidney cells (HEK293T; ATCC), were cultured in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal bovine serum (F1051; Sigma), 100 UI of penicillin, and 100 μg/mL of streptomycin (Wisent) at 37°C and 5% CO2. HCT116-ATF4 and HCT116-XBP1 cell lines were obtained by lentiviral infection of HCT116 cells. Briefly, 5 × 106 HEK293T cells were seeded on a 10-cm dish and transfected with the pLVx ATF4 mScarlet or XBP1 mNeonGreen transfer vectors as described, 22 and incubated for 24 h in 5 mL of fresh media 16 h posttransfection. Supernatant containing viral particles (5 mL) was filtered (0.45 μm filter), diluted 1:2 in fresh media, and supplemented with 8 μg/mL of hexadimethrine bromide (H9268; Sigma). The viral particles were used immediately to infect HCT116 cells (2 × 105), seeded 24 h before infection on a six-well plate (2 mL final volume). Cells were selected for host genome integration with 2 μg/mL of Puromycin (A1113803; Gibco). For PERK and IRE1 36-h monitoring experiments, 2.4 × 104 HeLa cells were seeded in a 96-well plate (flat bottom; Corning). HeLa cells were transfected 24 h after plating with 200 ng of either pLHCx ATF4 mScarlet or pLHCx XBP1 mNeonGreen plasmid DNA per well using XtremGeneHP (Roche) at a 3:1 ratio, then incubated for another 24 h. Cells were preincubated for 1 h before imaging with either DMSO or 2 μM thapsigargin. For PERK and IRE1 16-h monitoring experiments, 2 × 104 HCT116-ATF4 or HCT116-XBP1 cells were seeded 24 h before imaging on a PerkinElmer CellCarrier Ultra 384-well plate. Cells were preincubated for 30 min before imaging with either DMSO or 200 nM thapsigargin together with the indicated nuclear dye.
Image Acquisition
Image acquisition using Essen Bioscience Incucyte Zoom™ was performed with a 4 × NA 0.2 air objective. * After 1 h incubation with thapsigargin or DMSO, cells were imaged hourly for 36 h. Phase-contrast, GFP (400 ms exposure), and RFP (800 ms exposure) channels were used. Image acquisition using PerkinElmer Opera Phenix™ was performed in confocal mode with a 20 × NA 1.0 water immersion objective. † Time-course experiments were acquired for 16 h with one image every 30 min starting 30 min after thapsigargin addition. Unless otherwise stated, the channel configuration from Table 1 was used for image acquisition.
Image Acquisition Parameters
Image Analysis
For images collected with the Essen Bioscience IncuCyte Zoom, the percentage of Green/Red Object confluency was calculated using the Essen Bioscience software as the sum of the Green/Red objects area over the total image area. For images collected with the PerkinElmer Opera Phenix, quantification was performed on PerkinElmer Harmony® software. Segmentation was performed on brightfield images. Briefly, cell confluency mask was determined from collected images using a trainable texture classifier included in the “Find texture” tool of Harmony. This confluency mask was split into individual regions of interest (ROIs) using the “Find cells” tool of Harmony. Mean fluorescence values from each ROI were exported from Harmony and analyzed separately using a custom Python pipeline. Cells were determined as ATF4 or XBP1 positive when the mean ROI fluorescence intensity exceeded a threshold value determined using the following algorithm:
For each THR value in range (0–2,000):
where F_mean is the mean fluorescence intensity per ROI, THR is the variable threshold cutoff, T 0 and T 32 correspond, respectively, to the first and last scans of the plate on the Opera Phenix, R max the maximum observed value of R, and THRoptimal the value of THR corresponding to R max. Calibration was performed on a well containing unstained cells incubated with 200 nM thapsigargin, for each independent experiment, to scale the threshold value and maximize dynamic range between T 0 and T 32.
Statistical Analysis
Statistical significance between two groups was evaluated by unpaired Welsh-corrected Student's t-test.
Results
Construction of Improved PERK and IRE1α Pathways Sensors
Based on previous work in the literature, 16,18 the PERK activity sensor was built using atf4 5′ UTR region DNA sequence, where ORF1 and ORF2 are used as translation initiation in nonstressful conditions. However, phosphorylated eIF2α triggers translation initiation at ORF3 under ER stress. In the present construct, the fusion of mScarlet-I and c-myc NLS coding sequence in frame with the first 84 nucleotides of ORF3 allows the dynamic detection of ATF4 translation by quantifying fluorescence emission at 594 nm, which is indicative of PERK pathway activity (Fig. 1A). The IRE1 activity sensor was built using the 410–633 nucleotide sequence of the xbp1 cDNA, containing a 26 bp intron that is spliced in response to ER stress, by IRE1 RNase activity. The removal of the 26 bp intron leads to a frame shift that triggers translation of mNeonGreen fused with the c-myc NLS sequence, allowing dynamic detection of xbp1 splicing by quantifying fluorescence emission at 517 nm, hence IRE1 pathway activity (Fig. 1B).

Improved PERK and IRE1 sensors for detection of ER stress. Schematic of the sensors used:
Detection of ER Stress in HeLa Cells by Transfection of mScarlet ATF4 and mNeonGreen XBP1 Sensors
To test the activity of these new sensors, we cloned the encoding DNA sequences into the mammalian expression vector pLHCx and transfected them in HeLa cells. We performed western blot detection to verify the expression of the fusion proteins corresponding to the sensors, which was correlated to a known UPR response gene (CHOP), in the presence of tharpsigargin, an inhibitor of the SERCA Ca2+ pump at the ER membrane and known ER stress inducer (Fig. 1C, D). Interestingly, the expression of the HA-tagged fusion proteins for the ATF4 and XBP1 sensors was correlated to CHOP expression, although with a delay corresponding to ATF4-induced CHOP transcription. Therefore, both ATF4 and XBP1 sensors seem to elicit a faster response time and stronger signal compared with the detection of a standard UPR marker. Using an Essen Bioscience IncuCyte Zoom, we recorded successfully thapsigargin-induced ATF4 translation and xbp1 splicing dynamic over a 36-h time course and observed that the response peak was reached at 16 h for both sensors (Fig. 1E–G). However the day-to-day reproducibility of this assay was poor, as transient transfection introduced an increased variability in expression. As a result, data were not statistically significant for n = 3 experiments (Fig. 1H).
Single-Cell Detection of ATF4 Translation and xbp1 Splicing Using HCT116-ATF4 and HCT116-XBP1 Cell Lines
To eliminate the poor reproducibility due to transient transfection of the sensors, the HCT116 cell line was used for the stable expression of ATF4 mScarlet and XBP1 mNeonGreen by viral infection. We used a PerkinElmer Opera Phenix high-content imaging system (Table 1) to record the response to ER stress induced by thapsigargin in both HCT116-ATF4 and HCT116-XBP1 cell lines (Fig. 2A). For quantification, cell ROIs were extracted from the brightfield channel after segmentation with PerkinElmer Harmony software, enabling analysis of individual cells (Fig. 2B). Cells were scored positive for ATF4 translation (ATF4 positive) or xbp1 splicing (XBP1 positive) when the ROI mean fluorescence values were above a threshold value, determined with an adaptive method. As shown in Figure 2C and F, the dynamic responses of both ATF4 translation and xbp1 splicing in thapsigargin-treated cells were found to have a good dynamic range and remarkably high reproducibility for a cell-based assay.
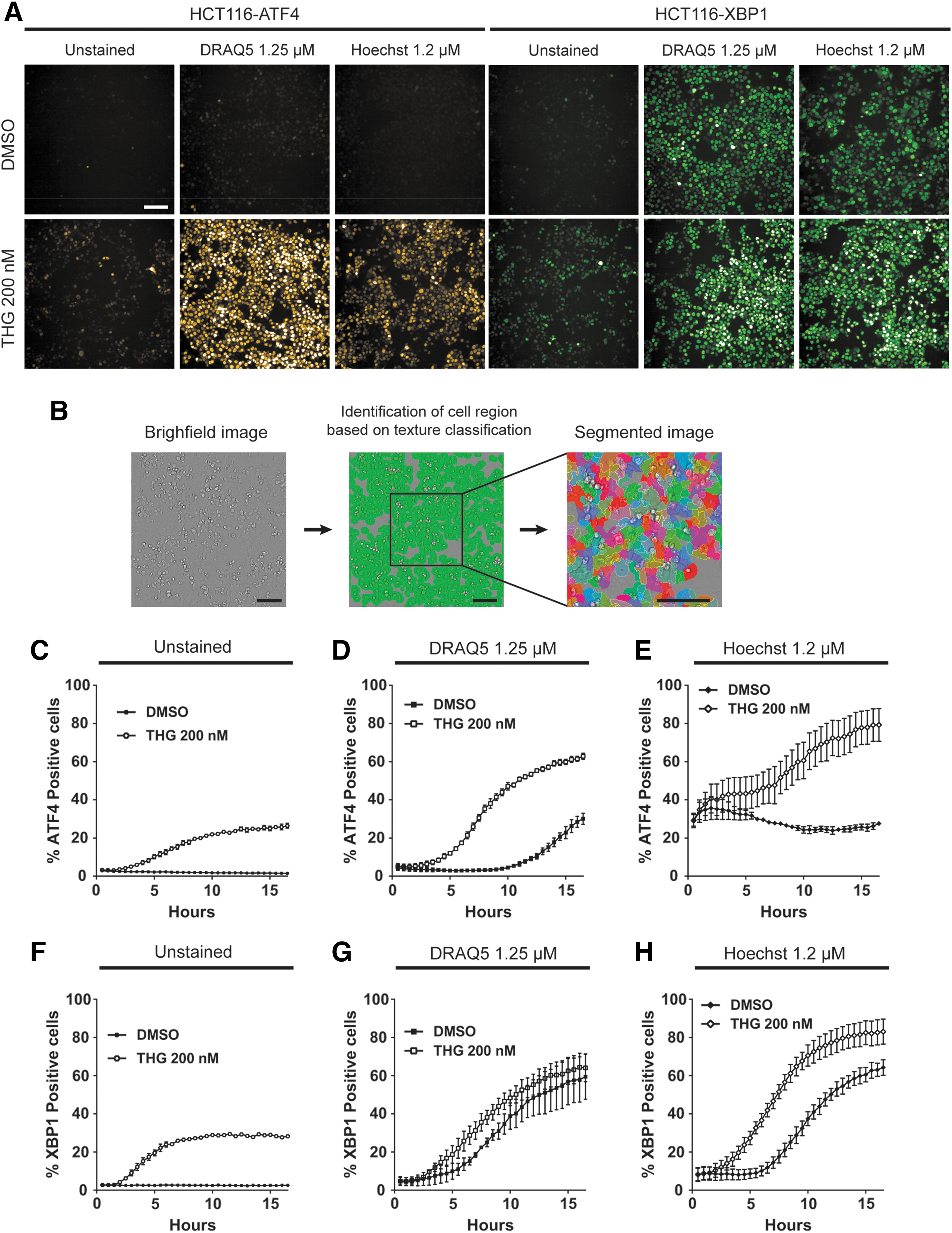
Dynamic response of ATF4 translation and xbp1 splicing at the single-cell level in mammalian cells is altered by nuclear stains DRAQ5 and Hoechst 33342.
Dynamic Response of ATF4 Translation and xbp1 Splicing Is Altered by Nuclear Stains DRAQ5 and Hoechst 33342
To enable direct application with other cell imaging platforms we substituted the brightfield-based segmentation with nuclei detection using either the far red nuclear stain DRAQ5 or blue Hoechst 33342. However, we observed that adding these nuclear dyes has a strong effect on both the ATF4 (Fig. 2D, E) and XBP1 (Fig. 2G, H) responses in thapsigargin-treated cells, increasing the number of positive cells. Furthermore, DRAQ5 elicited a modest ATF4 response in DMSO-treated control cells, but activated XBP1 pathway with almost the same kinetics as tharpsigargin (Fig. 2D, G). Hoechst 33342 had a very different effect than DRAQ5, with a strong initial activation of the ATF4 response, but almost no further increase in positive cells over the 16-h time course. Furthermore, Hoechst 33342 elicited a slightly higher magnitude and faster XBP1 response in thapsigargin-treated cells compared with the DMSO controls. Therefore, we conclude that brightfield-based segmentation is preferable to nuclear dye-based segmentation whenever ER stress responses may affect the biological readout of an assay in mammalian cells.
Alteration of ATF4 Translation and xbp1 Splicing Dynamic Response by Hoechst 33342 Is Dependent on Dye Concentration
To test whether the effect of Hoechst 33342 on ATF4 and XBP1 induction was dependent on the dye concentration, we examined concentrations of 0.18 and 0.036 μM. We did not perform further dilution of DRAQ5, as we observed that at lower concentrations dye retention within the nucleus was poor over a 16-h time course. As shown in Figure 3A–F the shape of the ATF4 and XBP1 response curves at 0.18 and 0.036 μM Hoechst 33342 were similar to the unstained cells in Figure 2C and F, whereas nuclei were still stained at those concentrations. Nevertheless, the background signal from ATF4 was higher at 0.18 μM Hoechst. Thus, as long as appropriate controls are included, Hoechst 33342 can be used at a concentration ranging from 0.18 to 0.036 μM for assays in which nuclei staining is required.

Alterations of ATF4 translation and xbp1 splicing responses by Hoechst 33342 is concentration dependent. ATF4 translation was quantified in HCT116-ATF4 cells over a 16-h time course in response to DMSO or 200 nM of thapsigargin in cells stained for 30 min with 1.2 μM
Differential ATF4 Translation and xbp1 Splicing Dynamic Responses Caused by Hoechst 33342 and DRAQ5 Are Partially Due to Dye-Mediated Phototoxicity
As excitation of fluorescent proteins and dyes has been linked with the generation of reactive oxygen species that are toxic for cells, 23 we investigated whether exposure to light from the excitation laser for DRAQ5 (640 nm) and Hoechst 33342 (375 nm) could be responsible for the toxicity observed. Indeed, turning off the 640 and 375 nm lasers abolished activation of the ATF4 pathway in DMSO-treated cells incubated with DRAQ5 or Hoechst 33342 (Fig. 4A–C), although these dyes still had a synergistic effect to potentiate thapsigargin activity as observed in Figure 4D–F. In contrast, the shape of the XBP1 response curve was not affected by turning down the 640 and 375 nm excitation lasers, although DRAQ5 and Hoechst 33342 excitation increased the total number of positive cells (Figs. 2F–H and 4D–F). Taken together, we conclude that ATF4 and XBP1 pathways' activation might be differentially sensitive to phototoxicity and that DRAQ5 and Hoechst 33342 effects on ER stress are only partially mediated by the excitation light source used for imaging.
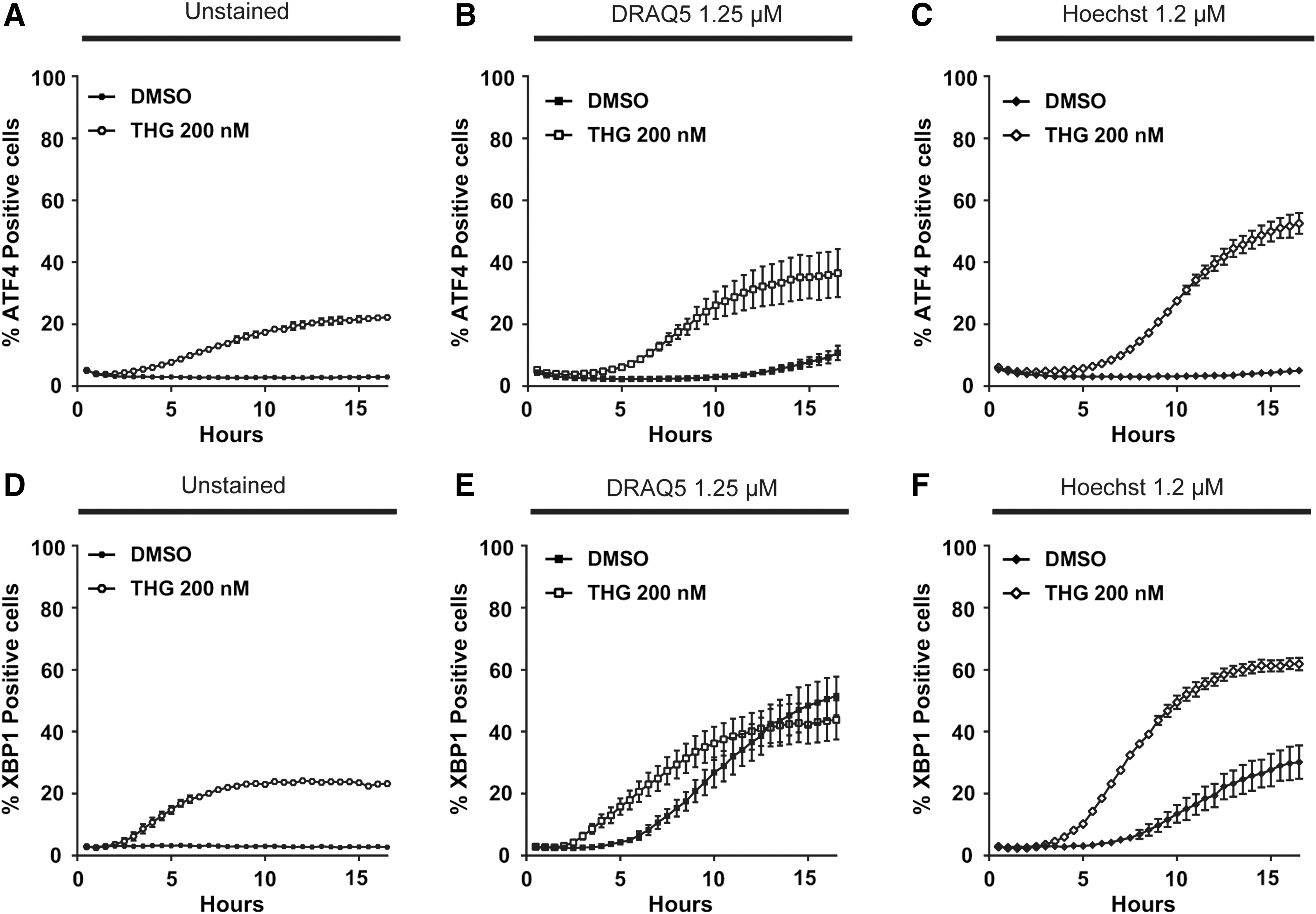
Differential ATF4 translation and xbp1 splicing dynamic response caused by Hoechst 33342 and DRAQ5 is partially due to dye-mediated phototoxicity. Opera Phenix™ 375 and 640 nm excitation laser intensities were decreased to quantify ATF4 translation in HCT116-ATF4 cells over a 16-h time course in response to DMSO or 200 nM of thapsigargin in cells without nuclear stain
A Multiplex Assay ER Stress Assay to Detect Simultaneously ATF4 Translation and xbp1 Splicing Dynamic Response in the Same Wells
Having optimized the sensors and the analysis method, we multiplexed the assay readout to record ATF4 translation and xbp1 splicing dynamic response in the same wells simultaneously. For that purpose, we seeded 384-well plates with a 1:1 mixture of HCT116-ATF4 and HCT116-XBP1 cells. Expressing the sensors in different cells rather than coexpression in a single cell leaves additional channels for other experimental readouts necessary to use the sensors in studies not focused entirely on stress responses. As detailed in Table 2, multiplexing enabled reproduction of the results obtained in Figure 4, where cell lines were seeded in separate wells, in half the acquisition time and number of wells (Fig. 5A–F). As expected, the number of ATF4- and XBP1-positive cells was halved, reducing the signal-to-background ratio for cells without nuclear stain, although reproducibility remained good. Therefore, multiplexing of the ATF4 and XBP1 detection can halve the reagent costs and more than double the speed of an assay at the cost of somewhat reduced sensitivity and slightly decreased reproducibility.
High-Throughput Multiplex Endoplasmic Reticulum-Stress Assay
1. Three hundred eighty-four-well CellCarrier Ultra 384 flat bottom plates (PerkinElmer) were used.
2. Verify that the plate level in the incubator is perfectly horizontal for the cells to settle nicely at the bottom of the plate.
3. Use 6 × drug and/or dye mix of ease of use. Save at least one well, where only thapsigargin is added at 200 nM final concentration to perform threshold calibration in the subsequent image analysis.
4. Verify that the focusing on the cells is adequate, as well as sufficient water is provided for the PerkinElmer Opera Phenix water immersion objective. Use confocal mode for the image acquisition.
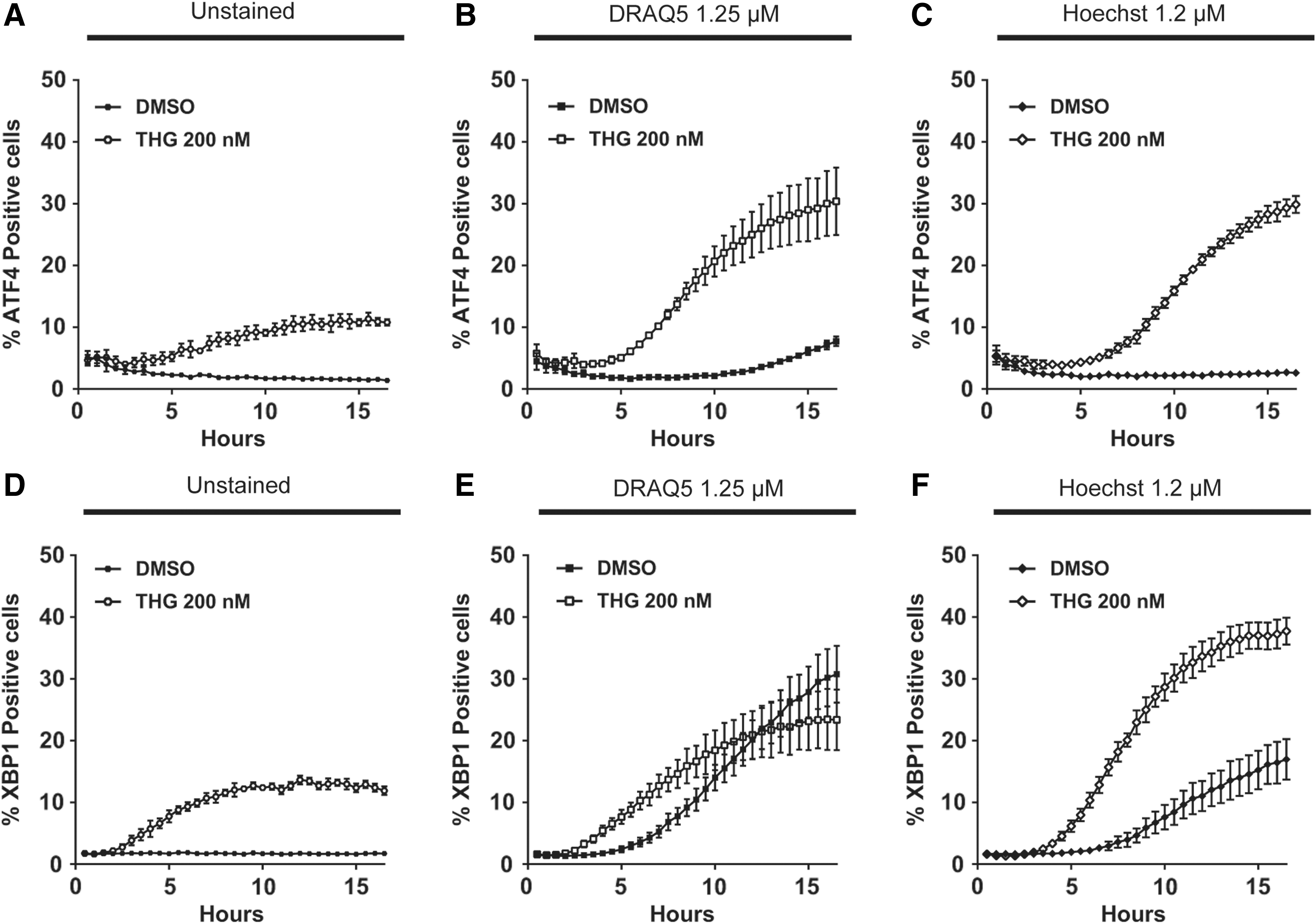
Multiplex assay ER stress assay to detect ATF4 translation and xbp1 splicing simultaneously in the same wells. HCT116-ATF4 and HCT116-XBP1 cells mixed at a 1:1 ratio in the same wells were imaged using a PerkinElmer Opera Phenix with the minimum required 375 and 640 nm excitation laser intensities, and emitted fluorescence intensity in the 488 and 561 nm channels was detected simultaneously over a 16-h time course in response to DMSO or 200 nM of thapsigargin. ATF4 translation was quantified in cells without nuclear stain
Discussion
Despite the involvement of the ER stress and the UPR pathways in numerous pathologies, efficient high-content screening detection methods have been lacking. The novel dual reporter system we validate in this study can accurately measure the early kinetic responses of the PERK and IRE1α pathways critical to determining cell fate in stressful conditions. 18
The use of the latest generation of fluorescent proteins, with a short folding time, enables direct recording of early UPR responses and improved performance compared with previous standard methods, such as western blot. The addition of a NLS sequence to the fluorescence protein fusions increased the specificity of the signal over previously reported constructs that resulted in a diffuse fluorescence signal across the cytosol. 16,18 Lentiviral-mediated integration of an xbp1 splicing-sensitive cassette, contrary to homologous recombination techniques used to generate a transgenic mouse model with an xbp1 splicing reporter, substantially reduced variability between replicates. Lentiviral transduction may allow use of our dual reporter system in many cell lines as well as primary cells that cannot be readily transfected.
Furthermore, single-cell-level analysis brought a higher level of accuracy into kinetic measurements of ER stress activation. However, obtaining single-cell data requires segmentation of images to identify nuclear areas, for analysis. While generally convenient, nuclear dyes should be avoided for time-course experiments, even without dye excitation, ATF4 and XBP1 pathways were affected by the dyes. Brightfield-based segmentation offers the best compromise for good signal-to-background ratio and reproducibility, but selection of alternative dyes might be considered.
Furthermore, we demonstrated that this dual ATF4 and XBP1 sensor system is scalable and displays sufficient reproducibility for high-throughput screening applications. Using mixed population cells containing either the ATF4 or XBP1 sensors should enable a further increased throughput and decreased cost in the case of a large screen. However, multiplexing can decrease the assay sensitivity, especially for detecting ATF4 translation. Finally, our simplified detection method for UPR should provide the tools required to incorporate stress responses into a variety of screens as well as to better understand the signaling pathways and to discover new therapies for diseases and conditions involving ER stress.
Footnotes
Acknowledgments
The authors thank Stéphane Borel for technical assistance. This project was supported by grant FDN143312 from the Canadian Institutes of Health Research (CIHR) to D.W.A. A.N. is supported by the French Fondation pour la Recherche Medicale (FRM) grant SPE20170839112. D.W.A. holds the Tier 1 Canada Research Chair in Membrane Biogenesis.
Disclosure Statement
No competing financial interests exist.