Turning Lemons into Lemonade Part I: Applications of Colloidal Drug Aggregates
Ganesh AN, Aman A, Logie J, Barthel BL, Cogan P, Al-awar R, Koch TH, Shoichet BK, Shoichet MS. Colloidal drug aggregate stability in high serum conditions and pharmacokinetic consequence. ACS Chem Biol 2019;14:751 − 757.
Abstract: Colloidal drug aggregates have been a nuisance in drug screening, yet, because they inherently comprise drug-rich particles, they may be useful in vivo if issues of stability can be addressed. As the first step toward answering this question, we optimized colloidal drug aggregate formulations using a fluorescence-based assay to study fulvestrant colloidal formation and stability in high (90%) serum conditions in vitro. We show, for the first time, that the critical aggregation concentration of fulvestrant depends on media composition and increases with serum concentration. Excipients, such as polysorbate 80, stabilize fulvestrant colloids in 90% serum in vitro for over 48 h. Using fulvestrant and an investigational pro-drug, pentyloxycarbonyl-(p-aminobenzyl) doxazolidinylcarbamate (PPD), as proof-of concept colloidal formulations, we demonstrate that the in vivo plasma half-life for stabilized colloids is greater than their respective monomeric forms. These studies demonstrate the potential of turning the nuisance of colloidal drug aggregation into an opportunity for drug-rich formulations.
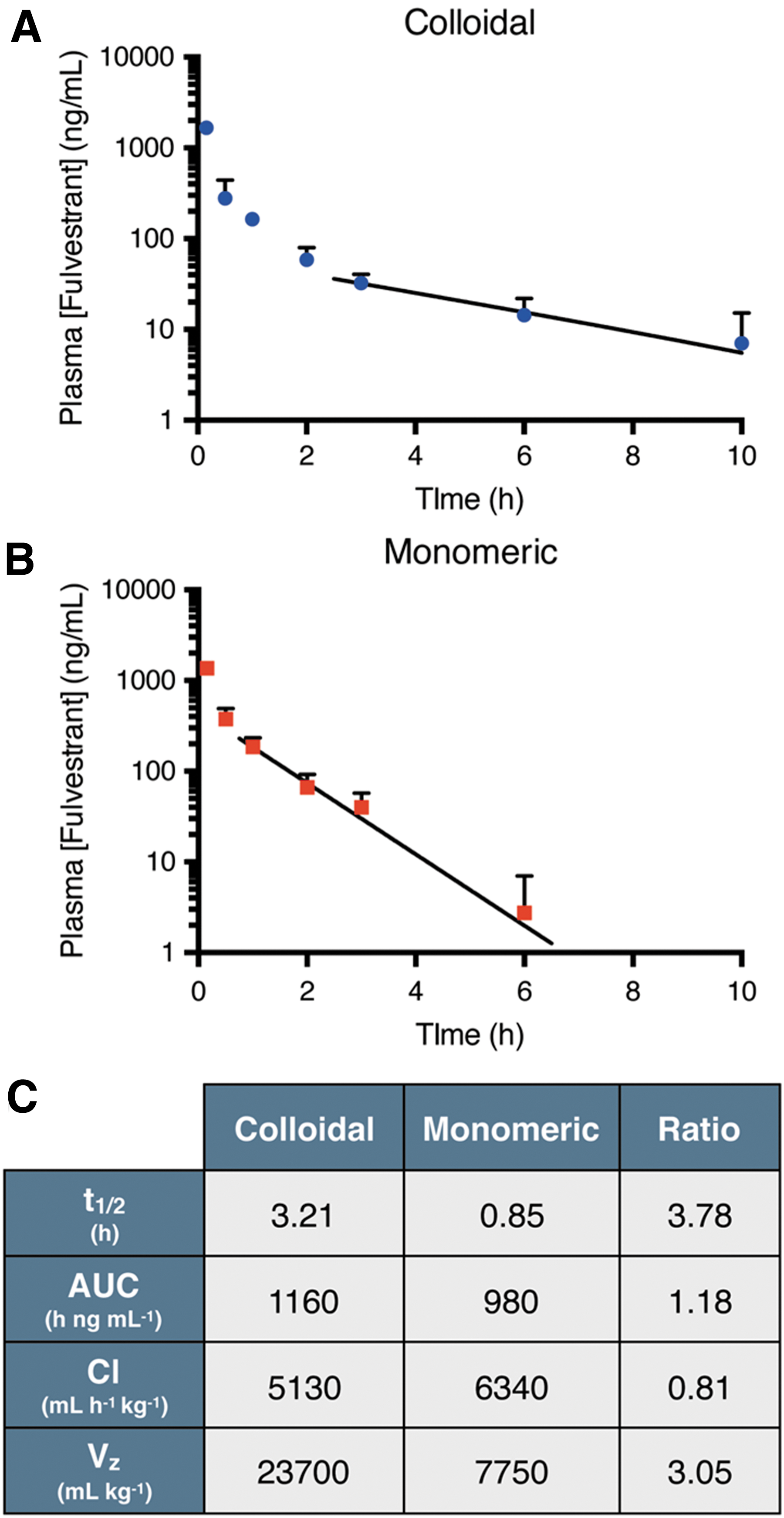
Commentary:Assay interference due to compound aggregation is a common mechanism among hits derived from HTS efforts. The colloidal particles that form once a compound is above its critical aggregation concentration (CAC) can lead to sequestration of assay components and attenuation of the assay signal. Previous work has also shown that compound colloids can form in typical cell culture media containing 10% serum, which limits the activity of compounds at high compound concentrations (see “Colloidal Pharmacology” ADDT literature review April 1, 2014; ACS Chem Biol 2014;9:777–784), leading to U-shaped concentration–response curves. Therefore, methods to flag activity due to compound aggregation and triage such compounds from hit lists have been developed and are often a primary concern when prioritizing compounds for follow-up activities (see the Assay Guidance Manual chapter;https://www.ncbi.nlm.nih.gov/books/NBK442297/). However, compound colloids can contain up to 108 molecules and as such could be useful for drug formulation if such drug-rich particles could be stabilized. Colloid formation has been observed in certain drugs, which affects the bioavailability, exposure, and distribution of the drug. This paper investigates the stability of colloidal particles under high serum conditions, which mimics in vivo conditions using a FRET reporter assay. The FRET assay employs a pair of cholesteryl BODIPY dyes excited at 490 nm with emission at 575 nm that are sequestered into the compound colloid, resulting in increased fluorescence upon colloid formation. One drug studied was the estrogen antagonist fulvestrant, a 3-hydroxy steroid. High serum concentrations (e.g., 90%) was found to increase the CAC and addition of the nonionic surfactant polysorbate 80 (UP80) at 0.1% was found to stabilize the fulvestrant colloids for 48 h in the presence of 90% serum. Comparing the pharmacokinetics mice dosed intravenously with either detergent solubilized (monomeric) fulvestrant or UP80 stabilized fulvestrant colloids shows a prolonged half-life and increased volume of distribution for the colloids (seefigure). The increased volume of distribution may be due to reduced plasma–protein–drug binding. Examination of the investigational anthracycline prodrug pentyloxycarbonyl-(p-aminobenzyl) doxazolidinylcarbamate (PPD) showed similar increases in half-life due to decreased drug clearance and increased drug exposure. The PPD colloids likely limit the metabolism by plasma carboxylesterase activity, which reduces the clearance of the drug. The FRET assay described here should be useful to study aggregation in vitro and formulation of serum-stable drug colloids can be used to manipulate the pharmacokinetics of drugs. Contributed by Doug Auld.
Pharmacokinetic profile of fulvestrant after intravenous administration. Plasma concentration of fulvestrant (initial dose, ID = 6 mg/kg, formulated at 1250 μM) administered as (A) stable colloids (0.03% UP80) or (B) monomer (5% UP80). Trend line denotes exponential decay fitting of lambda elimination phase. (C) Pharmacokinetic parameters of fulvestrant show almost 4-fold increase in drug half-life with colloids compared to monomer (n = 3 − 6, mean + SD).
TURNING LEMONS INTO LEMONADE PART II
Donders EN, Ganesh AN, Torosyan H, Lak P, Shoichet BK, Shoichet MS. Triggered release enhances the cytotoxicity of stable colloidal drug aggregates. ACS Chem Biol 2019 Jun 25 [Epub ahead of print]; DOI: 10.1021/acschembio.9b00247.
Abstract: Chemotherapeutics that self-assemble into colloids haven limited efficacy above their critical aggregation concentration due to their inability to penetrate intact plasma membranes. Even when colloid uptake is promoted, issues with colloid escape from the endolysosomal pathway persist. By stabilizing acid-responsive lapatinib colloids through coaggregation with fulvestrant, and inclusion of transferrin, we demonstrate colloid internalization by cancer cells, where subsequent lapatinib ionization leads to endosomal leakage and increased cytotoxicity. These results demonstrate a strategy for triggered drug release from stable colloidal aggregates.
Commentary:Compounds that form colloids can limit the activity in cell-based assays when concentrations above the CAC are achieved. The present study shows that this can be remedied by protonation of acid-responsive drugs, inducing endocytosis, and intracellular release of the drug, which is triggered by endosomal disruption. A series of drugs that show pH-dependent formation of colloids (pKa >5) are studied. Stable colloids of the acid-responsive drug lapatinib were made by co-aggregating with fulvestrant at a 1:3 molar ratio of lapatinib/fulvestrant. Acidification of these particles showed increased release of lapatinib with decreasing pH, while free levels of the nonionizable-drug fulvestrant were unchanged. This supports that free lapatinib will be formed when the colloid is present within acidic intracellular compartments such as endosomes. To study intracellular uptake and release of these colloids, a transferrin-stabilized lapatinib/fulvestrant was formulated, which resulted in cellular uptake of the particle by endocytosis. The same FRET assay as described in the accompanying commentary was used to monitor the colloidal particles, and a membrane impermeable dye (7-aminoactinomycin D, 7-AAD) was also incorporated into the colloids, which upon uptake and subsequent release results in DNA binding and increased fluorescence of the nucleus. The transferrin stabilized colloids showed formation of punctate within cells, increased cytotoxicity, and increased DNA staining, which was not observed when stabilized colloids were used lacking transferrin (seefigure). This paper further demonstrates how colloidal drug particles can be applied to manipulate the activity drugs in cells. Contributed by Doug Auld.
Lapatinib/fulvestrant colloids are cytotoxic after endocytosis and subsequent endosome disruption. Colloidal drug aggregates were formulated as described in Table S3, with the addition of 2 μM 7-aminoactinomycin D (7-AAD) to monitor endosome disruption in those experiments. Colloids composed of 50 μM lapatinib and 150 μM fulvestrant and stabilized with transferrin resulted in greater (A) cytotoxicity and (B) endosomal disruption (measured by amount of nuclear dye) than either co-colloids stabilized with PLAC-PEG or colloids of fulvestrant alone (150 μM fulvestrant plus stabilizer) (n = 9 biological replicates and separate colloid formulations for the toxicity experiment and n = 3 for the endosome disruption experiment, mean ± SD, two-way ANOVA with Tukey's posthoc test, *p < 0.05, **p < 0.01, ****p < 0.0001). (C) Schematic describing how the membrane-impermeant nucleic acid stain 7-AAD was used to test for endosomal escape. (D) Nuclear 7-AAD fluorescence (red) is visible after treatment with lapatinib-fulvestrant colloids stabilized with transferrin, demonstrating its endosomal escape. (E) Permeabilized cells accumulate 7-AAD (red) in their nuclei as expected, whereas (F) live cells treated with 7-AAD alone do not. The presence of cells in each region of interest was verified using the transmission channel prior to capturing these images
From Antibody to a Small Molecule
van Dongen MJP, et al. A small-molecule fusion inhibitor of influenza virus is orally active in mice. Science 2019;363: DOI: 10.1126/science.aar6221.
Abstract: Recent characterization of broadly neutralizing antibodies (bnAbs) against influenza virus identified the conserved hemagglutinin (HA) stem as a target for development of universal vaccines and therapeutics. Although several stem bnAbs are being evaluated in clinical trials, antibodies are generally unsuited for oral delivery. Guided by structural knowledge of the interactions and mechanism of anti-stem bnAb CR6261, we selected and optimized small molecules that mimic the bnAb functionality. Our lead compound neutralizes influenza A group 1 viruses by inhibiting HA-mediated fusion in vitro, protects mice against lethal and sublethal influenza challenge after oral administration, and effectively neutralizes virus infection in reconstituted three-dimensional cell culture of fully differentiated human bronchial epithelial cells. Cocrystal structures with H1 and H5 Has reveal that the lead compound recapitulates the bnAb hotspot interactions.
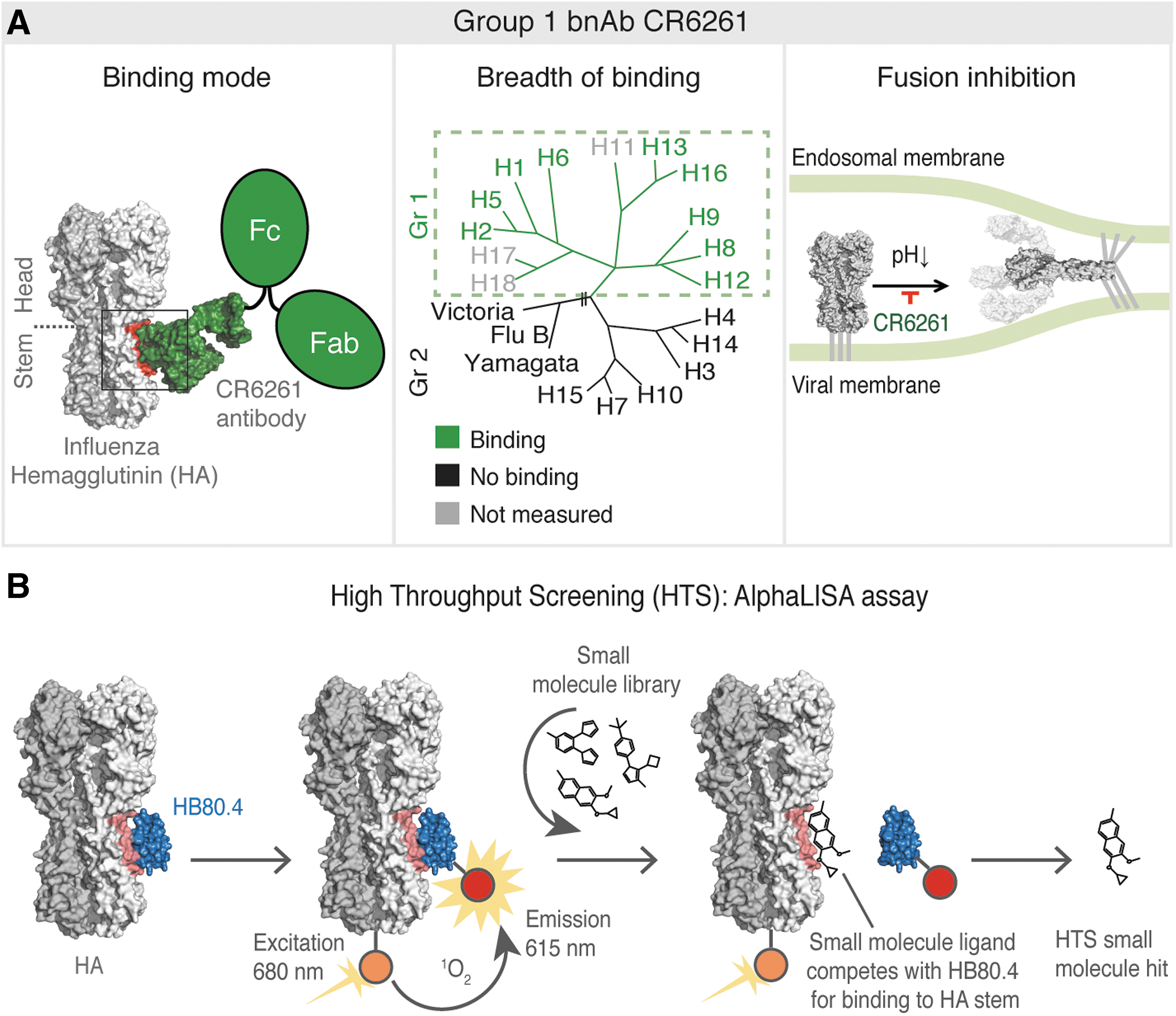
Commentary:Antibodies neutralizing viral entry/fusion have enjoyed a growing success, in part due to the fact that they can physically cover/block relatively large portions of the target proteins, a feat not easily accomplished by a small molecule. At the same time, antibody therapeutics are not well suited for an easy oral administration. In the present work, van Dongen et al. designed and traversed an exciting path from a broadly neutralizing anti-influenza antibody to an orally bioavailable small molecule that recapitulated the antibody's function. Starting from the broadly neutralizing antibody (bnAb) CR6261, the authors first used computational approaches to design a small synthetic protein named HB80.4 that mimicked the antibody's combining site and exhibited a similar fusion inhibition profile. This was done in order to avoid the antibody's avidity effect that would have been impossible to copy through a small molecule. With the new HB80.4 binder at hand, the team developed an AlphaLISA assay to screen for small molecules capable of displacing HB80.4 from its binding interaction with the stem portion of hemagglutinin (HA; seefigure). Out of approximately half a million compounds screened and 300 hits progressed to validation, a benzylpiperazine series was studied in more detail, ultimately leading to the generation of JNJ4796 as the lead molecule. JNJ4796 was examined in influenza models in mice where it displayed good oral bioavailability and efficacy, as expected based on the original bnAb mechanism of action. Overall, this study provides a good framework for how one can “rework” an antibody into a small molecule that phenocopies the original mechanism of action. Contributed by Anton Simeonov.
influenza antibody CR6261. (A) Binding mode, breadth of binding, and fusion inhibition profile of influenza HA stem–targeting antibody CR6261. The left panel shows the binding mode of CR6261 to influenza HA, with the HA trimer represented as a gray molecular surface with different shades for the different protomers, CR6261 in green with a molecular surface for the Fab interacting with the HA and a cartoon for the other Fab and Fc of the immunoglobulin G, and the binding epitope on the HA in red. The middle panel illustrates the HA phylogenetic tree, which shows the relationship between the 18 HA subtypes of influenza A virus (group 1 and 2) and the two lineages of influenza B viruses. The breadth of binding of the CR6261 to multiple group 1 subtypes is shown in green, light gray indicates where binding was not tested, and black indicates no binding. The right panel shows the mechanism of action of CR6261 to block the pH-induced HA conformational changes. (B) Our HTS utilized the AlphaLISA technology to identify small molecules targeting the CR6261 epitope. The CR6261-mimicking designed protein HB80.4 was used in the binding competition assay. HB80.4 is represented in blue and the CR6261 epitope on the HA in pink. Small molecules are indicated as illustrative structural formulae.
Cancer-Causing Carbohydrate
Engle DD, et al. The glycan CA19-9 promotes pancreatitis and pancreatic cancer in mice. Science 2019;364:1156–1162.
Abstract: Glycosylation alterations are indicative of tissue inflammation and neoplasia, but whether these alterations contribute to disease pathogenesis is largely unknown. To study the role of glycan changes in pancreatic disease, we inducibly expressed human fucosyltransferase 3 and β1,3-galactosyltransferase 5 in mice, reconstituting the glycan sialyl-Lewisa, also known as carbohydrate antigen 19-9 (CA19-9). Notably, CA19-9 expression in mice resulted in rapid and severe pancreatitis with hyperactivation of epidermal growth factor receptor (EGFR) signaling. Mechanistically, CA19-9 modification of the matricellular protein fibulin-3 increased its interaction with EGFR, and blockade of fibulin-3, EGFR ligands, or CA19-9 prevented EGFR hyperactivation in organoids. CA19-9–mediated pancreatitis was reversible and could be suppressed with CA19-9 antibodies. CA19-9 also cooperated with the KrasG12D oncogene to produce aggressive pancreatic cancer. These findings implicate CA19-9 in the etiology of pancreatitis and pancreatic cancer and nominate CA19-9 as a therapeutic target.
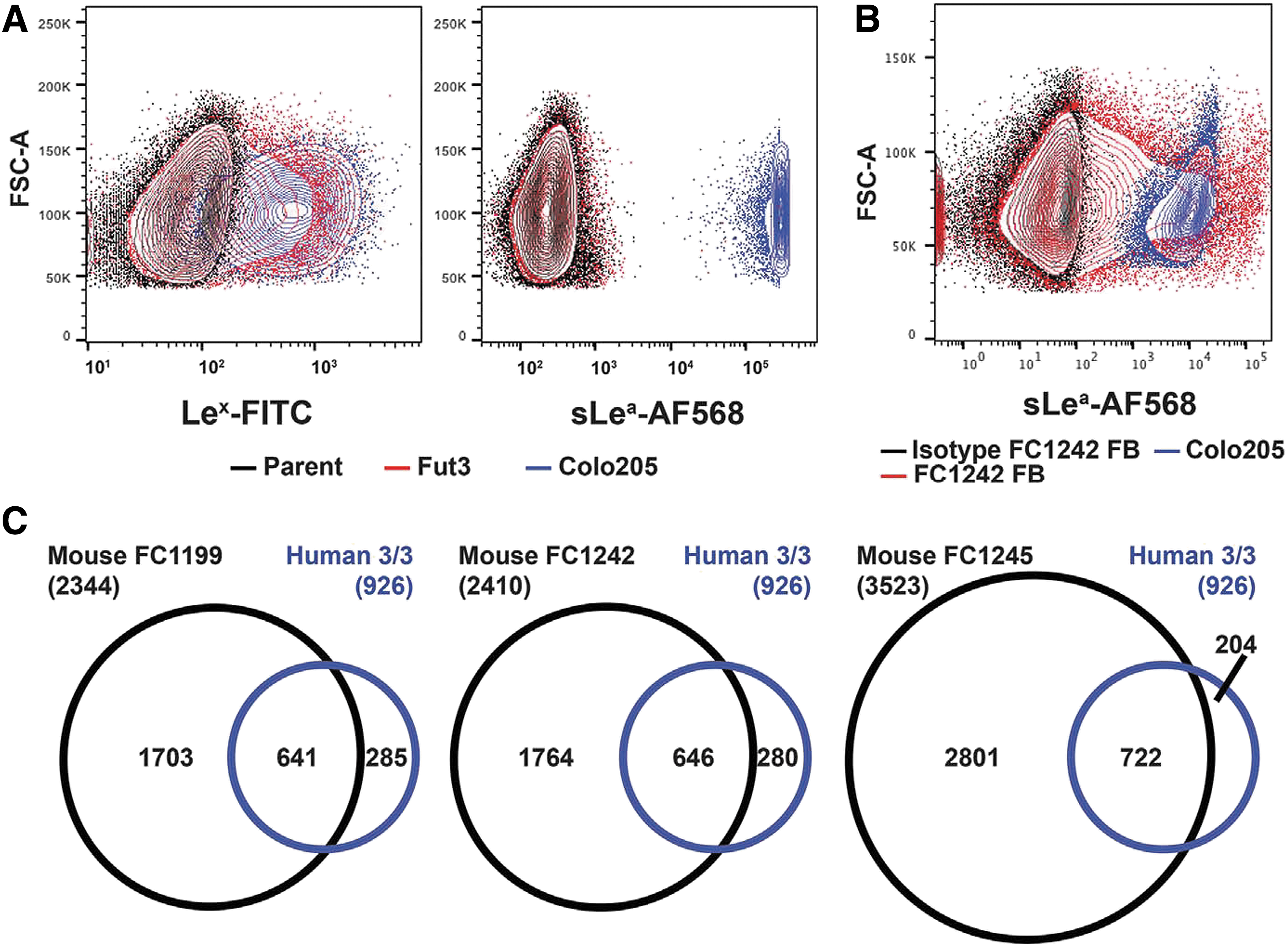
Commentary:Sialyl-Lewisa (sLea), also known as CA19-9, is a glycan carbohydrate modification on the surface of proteins, and over the years it has become associated with pancreatitis and pancreatic cancer. Indeed, diagnostic approaches to pancreatic cancer detection have incorporated assays for CA19-9. The present study reports on the potential of CA19-9 to promote the development of pancreatic cancer. To be able to study the potential causative effect of CA19-9, the team first needed to develop a method for the controlled overexpression of CA19-9 in mice. This was not a straightforward task because CA19-9 is not a typical single protein traditionally studied in the context of carcinogenesis. Rather, it is a posttranslational modification. As such, the authors needed to rewire the carbohydrate metabolism in the study animals in order to provide for the right carbohydrate precursors leading up to the CA19-9 modification (seefigure). This new mouse model allowed the comprehensive study of the effect of CA19-9 overexpression in mice. The team showed that increase of CA19-9 in mice triggers acute and chronic pancreatitis with further hyperactivation of epidermal growth factor receptor (EGFR) signaling and ultimately a pancreatic tumorigenesis. Conversely, treatment of mice in this model with two antibodies directed against CA19-9 reduced the EGFR hyperactivation and allowed for the tumor management. The authors noted that using erlotinib to target EGFR in this model was less effective than the use of anti-CA19-9 antibodies, presumably because the antibody is acting upstream of EGFR. Thus, this study “upgrades” the role of CA19-9 from a biomarker to a driving factor in pancreatic cancer, and argues for efforts to target CA19-9 for potential therapeutic benefit. Contributed by Anton Simeonov.
(B) CA19-9 flow cytometry of mouse PDAC cells stably and constitutively expressing FUT3 with β3GALT5 (FB) compared with the isotype control antibody. (C) Overlap between CA19-9 protein carriers identified in three out of three human PDAC cell lines (n = 926) with three independent mouse PDAC cell lines expressing FUT3 and β3GALT5.