
Abstract
Phospholipid biosynthesis begins with the acylation of glycerol 3-phosphate (G3P). In most Gram-positive bacteria including many pathogens, a membrane protein called PlsY is the only acyltransferase that catalyzes this essential step, making it a potential target for the development of antibiotics. A convenient enzymatic assay should facilitate such drug discovery activities. Previously, we developed a continuous assay by monitoring phosphate, one of the enzymatic product, using a fluorescently labeled phosphate binding protein in a bilayer environment called lipid cubic phase (LCP). However, some intrinsic characteristics of LCP, such as high viscosity, make the assay incompatible with common high-throughput liquid-handling platforms. Here, we adapted the assay by hosting PlsY in detergent micelles, enabling us to conduct the assay using standard multi-channel pipets in a high-throughput manner. With optimal enzyme loading, the reaction velocity was linear up to 30 min. PlsY showed Michaelis–Menten kinetics behavior in micelles with a V max of 57.5 μmol min−1 mg−1, and K m of 1.14 mM G3P and 6.2 μM acyl phosphate. The inhibitory product lysophosphatidic acid inhibited PlsY with the IC50 of 19 μM. The results principally demonstrated the feasibility of using the assay for high-throughput screening, and the protocol provided an encouraging starting point for further optimization and validation of the assay for automated platforms.
Introduction
The evolution of drug resistance in bacteria render the current antibiotics ineffective and outpace the discovery of new drugs, posing a serious threat to public health. 1 New antibiotics are urgently needed to fight such multidrug-resistant bacteria such as methicillin-resistant Staphylococcus aureus. 2 Phospholipids are a major and essential part of the membrane. Thus, enzymes in the phospholipid biosynthesis pathway are potential targets for inhibition.
The phospholipid biosynthesis pathway commences with the acylation of glycerol 3-phosphate (G3P), forming lysophosphatidic acid (lysoPA), which undergoes a second acylation step to produce phosphatidic acid. Two types of G3P acyltransferases with no shared sequence homology exist. The PlsB type uses acyl coenzyme A as the acyl donor and produces free coenzyme A as the by-product. The PlsY type uses the unusual acyl phosphate (acylP) as the acyl donor, and produces inorganic phosphate (Pi) as the by-product. 3,4 In Gram-positive bacteria like Staphylococcus, PlsY is the only G3P acyltransferase, and the disruption of the gene was reported to be lethal. 5,6 Importantly, PlsY has no mammalian homologs, making it an attractive drug target. 4,7,8 Substrate analogs had been designed and tested as potential PlsY inhibitors to suppress the growth of pathogens. 7,8
PlsY is relatively small (∼20 kDa) and remarkably hydrophobic; it spans membrane seven times and 80% of its residues are buried in the membrane. 9,10 Recently, we crystallized PlsY and solved several high-resolution structures of PlsY, collectively with all substrates/products bound, revealing a V-shaped active site and its unique catalytic mechanism. 9,11 With atomic details of the active site architecture, it is now possible to screen small molecules in silico, an approach that generally scores millions of compounds and generates dozens to hundreds of candidates for experimental evaluation. 12 Alternatively, small-molecule libraries can be screened for hits that inhibit PlsY. 13 A mid-to-high-throughput assay would facilitate both processes.
Here, we reviewed the current assays and reasoned that they were unsuitable for high-throughput screening. Accordingly, we modified an existing coupled assay so that the assay could be performed in a high-throughput multi-well format and in a liquid-handling friendly manner. Using this assay, we characterized PlsY in detergent micelles for the first time by determining the kinetic constants and measuring the IC50 of the product inhibitor lysoPA. The assay could be utilized for mid-scale screening of compounds using standard laboratory pipets and had the potential for automated platforms for high-throughput applications.
Materials and Methods
Materials
Detergents dodecyl maltoside (DDM, cat. no. D310S) and lauryl maltose neopentyl glycol (LMNG, cat. no. NG310) were obtained from Anatrace (Maumee, OH). AcylP was synthesized as described below. G3P (cat. no. G6501) was purchased from Sigma (St. Louis, MO). lysoPA (cat. no. 857123P) was sourced from Avanti (Alabaster, AL). N-[2-(1-maleimidyl)ethyl]-7-(diethylamino)coumarin-3-carboxamide (MDCC, cat. no. D10253) was purchased from Thermo Fisher (Waltham, MA). Other buffer and salts were obtained from Sigma.
Purification of PlsY
The purification of PlsY from Aquifex aeolicus was carried out as described. 9 Briefly, the enzyme was recombinantly expressed in Escherichia coli BL21 (DE3) cells as a fusion protein with a C-terminal green fluorescent protein (GFP) and an 8 × His tag. For purification, all procedures were carried out at 4°C unless specified otherwise. Cell lysates were centrifuged at 20,000 g for 30 min to remove cell debris. The membrane fraction was spun down by centrifugation at 150,000 g for 1.5 h. Membrane proteins were solubilized with 1% (w/v) of DDM for 1.5 h. The mix was heated at 65°C for 10 min, and centrifuged at 48,000 g for 1 h to remove insoluble materials and precipitated proteins. The supernatant containing His-tagged PlsY-GFP fusion protein was incubated with nickel nitrilotriacetic acid (Ni-NTA) resin for 2 h. The resin was washed with 10 and 45 mM imidazole to remove contaminants. His-tagged PlsY-GFP fusion protein was eluted with 0.25 M imidazole, desalted, and digested with home-purified 3C protease (see below) to generate tag-free PlsY. The mixture was incubated with Ni-NTA resin again to separate the tag-free PlsY from the His-tagged GFP and the His-tagged 3C protease. PlsY, collected as the flow-through fraction of the second Ni-affinity chromatography step, was concentrated to 20 mg mL−1, flash frozen in liquid nitrogen, and stored at −80°C until use.
Purification of the 3C Protease
The detailed procedure for the purification of His-tagged 3C protease was also published previously. 9 Briefly, the protease was expressed in E. coli BL21 (DE3) cells as a fusion protein, from the N- to C-terminus, as the glutathione S-transferase (GST) tag, 3C protease cleavage site (LEVLFQ^GP), an 8 × His tag, and the 3C protease. During the expression, the GST tag is released by the protease itself, yielding an N-terminally His-tagged protease. The protease was purified using Ni-NTA resin. The final product was stored at −80°C in 0.2 mM tris(2-carboxyethyl)phosphine, 20 mM Tris HCl pH 8.0.
Purification of Phosphate Binding Protein
Phosphate binding protein (PBP) (residue Glu26-Tyr346 with the mutation A197C) 9,14 was expressed in E. coli with a C-terminal His-tag. Cells were lysed and centrifuged at 20,000 g for 20 min. The clarified lysate was incubated with Ni-NTA resin for 2 h to allow batch-binding. The resin was then washed extensively with 10 mM Tris-HCl pH 8.0, followed by washes with 40 mM imidazole in the Tris buffer. PBP was eluted with 250 mM of imidazole in the Tris buffer, desalted, and loaded onto a 5-mL HiTrap Q Sepharose FF column pre-equilibrated with the Tris buffer. PBP was eluted using 40 mL of 0–100 mM NaCl in the Tris buffer at a flow rate of 1 mL min−1, concentrated to 32.5 mg mL−1, flash frozen in liquid nitrogen, and stored at −80°C.
Fluorescent Labeling of PBP
The labeling of PBP with the thio-reactive MDCC was carried out as described. 14 Residual Pi was removed in a 4-mL reaction mix containing 13 mg of PBP, 50 μg mL−1 of purine nucleoside phosphorylase (PNPase), 0.2 mM of 7-methylguanosine (MEG), and 10 mM Tris-HCl pH 8.0 for 30 min at 20°C. PBP was then labeled with 0.15 mM of MDCC for 2 h at 20°C. Unreacted MDCC was removed by gel filtration on a Superdex 200 10/300 GL column pre-equilibrated with 10 mM Tris HCl pH 8.0. Labeled and free PBP were separated on a 5-mL HiTrap Q Sepharose FF column with 200 mL of 0–250 mM NaCl gradient wash at 1 mL min−1. The MDCC-PBP concentration was calculated using the following formula: ([A280, 1 cm−A430, 1 cm × 0.164]/61,656 M−1). 14 The MDCC-PBP was concentrated to 3.7 mg mL−1 (0.1 mM), flash frozen in liquid nitrogen and stored at −80°C.
Purification of the PNPase
The PNPase from E. coli was recombinantly expressed in E. coli BL21 (DE3) cells with an N-terminal 8 × His-tag and purified as described. 8 Cell lysates were clarified by centrifugation at 48,000 g for 50 min at 4°C. Ni-NTA resin was added to the supernatant and the mix was gently stirred for 1 h. After the batch-binding, the resin was washed with 35 mM imidazole in Buffer A containing 0.2 mM EDTA, 10% (v/v) glycerol, 100 mM Tris HCl pH 7.5. The protein was eluted with 0.25 M imidazole, desalted, and further purified by anion exchange on a 5-mL HiTrap Q Sepharose FF column. The enzyme was eluted with 100 mL of 0–500 mM NaCl in Buffer A.
Chemical Synthesis of acylP
Palmitoyl phosphate was synthesized as described. 9,15,16 Briefly, 3.76 g Ag3PO4 and 2.08 g of 92% (w/v) H3PO4 were mixed and ground with 40 mL of cold ether in a morter inside an ice box. The suspension was transferred to a sealed bottle and stirred on ice for 1.5–2 h. Palmitic chloride (5 mL) dissolved in 20 mL of cold ether was added drop-wise to the suspension. The mixture was ground, stirred on ice for another 20 min, and centrifuged at 4,000 g for 10 min at 4°C. The supernatant and the washes were combined, and added to a round-bottom flask that is attached to a rotary evaporator for removal of solvents. To the dried substance, warm benzene (60°C, 100 mL) was added, resulting in a turbid suspension. The suspension was passed through four layers of filter paper, and the filtrate was incubated at 6–8°C for 4 h for crystallization of acylP. White crystals were harvested by filtration, washed with cold benzene, and dried in vacuo (0.07 mBar, −102°C). The identity of the product was confirmed using NMR and mass-spec analysis: 1 H NMR (dimethyl sulfoxide, 400 MHz), 0.85 (t, 3H, J = 6.4 Hz), 1.24 (s, 26H), 1.45–1.56 (m, 2H), 2.38 (t, 2H J = 7.2 Hz); Mass-Spec (electrospray ionization) m/z = 335 [M 2H]−. The final product was placed in a sealed tube with desiccator and stored at −80°C.
Measurement of Pi Levels in DDM and LMNG
DDM and LMNG stock solutions were prepared by dissolving them in MilliQ water to 10% (w/v) concentration. Detergents and MDCC-PBP were added to the buffer (150 mM NaCl, 50 mM Tris HCl pH 8.5) to final concentrations of 0.03% (w/v) and 4 μM, respectively. For standards, known amounts of Na2HPO4 were added to 4 μM MDCC-PBP, 150 mM NaCl, and 50 mM Tris HCl pH 8.5. Fifty microliters of solutions was transferred into a black half-area 96-well plate for fluorescence (excitation = 425 nm, emission = 466 nm) measurement using the “bottom-read” mode. Typically, 1 μM of Pi causes a 1,550-count change for the fluorescence of MDCC-PBP on our M5e plate reader (Molecular Devices). The amount of free Pi was then calculated based on the calibration curve and the dilution factors.
Removal of Free Pi in G3P
The sequestration of free Pi in G3P was carried out following our previous protocol. 9 The stock solution of G3P (1 M) was incubated with 50 μg mL−1 of PNPase, 5 mM MEG for 1 h at 20°C, protected from light. The mixture was then heated at 60°C for 10 min to inactivate PNPase. The level of free Pi in the G3P treated above was under detection limits of the MDCC-PBP probe.
Removal of Free Pi in acylP
AcylP (2.5 mg of powder) was added from the back of a Hamilton syringe (cat. no. 80930). Sixteen microliters of MilliQ water was added into a 100-μL Hamilton syringe from the front side. A second syringe filled with 24 μL of monoolein was then connected to the first syringe via a coupler. 17 Lipid cubic phase (LCP) was obtained by pushing the plungers back and forth for about 200 times. The LCP was injected into a 1.5-mL Eppendorf tube, soaked with buffer containing 150 mM NaCl and 50 mM Tris-HCl pH 8.5 for 5 min for five times, dissolved in 50 μL of dimethylsulfoxide (DMSO), aliquoted, and stored at −80°C until use. The contaminant Pi level was reduced to levels that were not detectable by the MDCC-PBP assay.
Activity Assay of PlsY in 96-Well Plates
To a half-area 96-well plate, 45 μL of assay buffer containing PlsY, acylP, and MDCC-PBP was added and incubated at 26°C. Five microliter of G3P was added to initiate the reaction. The fluorescence (emission 425 nm, excitation 466 nm) was read every 30 s for 1 h using the “bottom-read” mode. For the enzyme loading assay, G3P and acylP were kept at 20 mM and 40 μM, respectively; while PlsY was varied from 0 to 10 ng mL−1. For the K m determination of G3P, acylP was kept at 20 μM and the enzyme was at 5 ng mL−1, and G3P was varied from 0 to 10 mM. For the K m determination of acylP, G3P was at 10 mM and the enzyme was kept at 5 ng mL−1, and acylP was varied from 0 to 60 μM. To determine the IC50 of lysoPA, the concentrations for the enzyme, G3P, and acylP were 1 ng mL−1, 1.14 mM, and 6.2 μM, respectively. LysoPA was varied from 0 to 100 μM. Kinetic parameters were calculated using Prism.
Results and Discussion
Rationale for Assay Development
The conventional assay of G3P acyltransferases including PlsY monitors the transfer of radioactivity from the water-soluble phase to the organic phase as a result of the acylation of the radio-labeled G3P. 16,18 –20 Although the assay is very sensitive, it is not desirable for high-throughput screening, because it involves multiple steps such as repeated extraction and evaporation of solvents. In addition, it requires special reagents, equipment, and procedures for radioactive substances.
Recently, we developed an assay to characterize PlsY in LCP, a lipid bilayer environment,
9
by monitoring the fluorescence change of PBP labeled with MDCC in response to the release of the enzymatic product phosphate (Fig. 1A).
14
Although the assay had the advantage in providing kinetic information of PlsY in a bilayer mimetic, the intrinsic feature of LCP poses two practical hurdles for high-throughput screening. First, the highly viscous,
21
gel-like LCP is not compatible with liquid-handling robots in common high-throughput screening platforms. Second, the geometry of the LCP assay setup would limit the encounter between the membrane protein enzyme and inhibitors (Fig. 1B
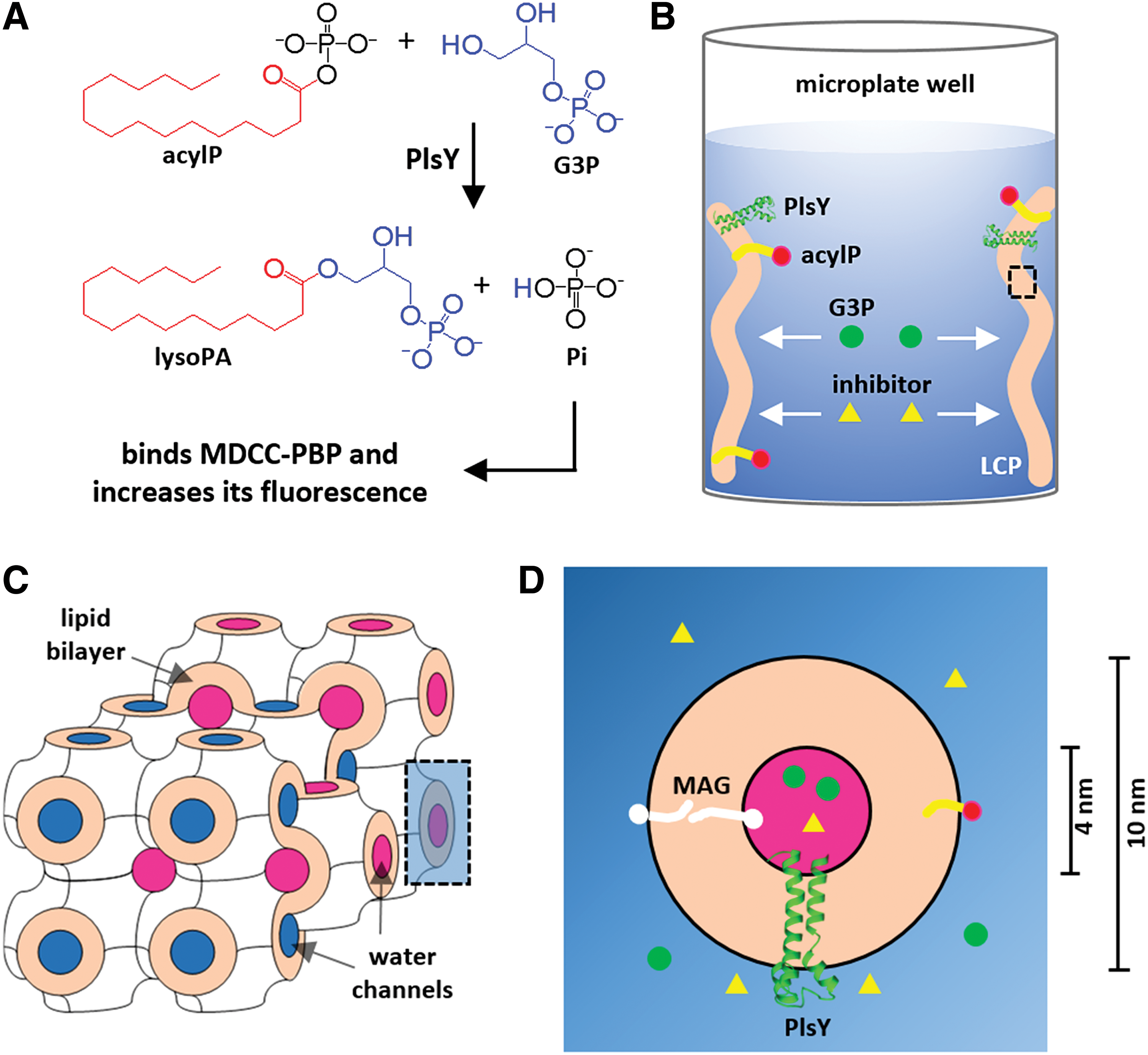
The LCP assay was reasoned to be unsuitable for high-throughput screening. The coupled assay monitors the production of Pi using the Pi-biosensor, MDCC-PBP
One might consider making LCP using PlsY preloaded with compounds to circumvent the diffusion issue. However, individual LCP samples would then be required for each compound, which is too time-demanding (20 min for each) to be feasible. In addition, much of the LCP material would be wasted because the minimum reliable volume for making an LCP sample is about 15–20 μL, while only 1 μL is needed for each inhibitor for initial screening. Taken together, we concluded that the LCP assay was unsuitable for inhibitor screening.
Removing Contaminant Pi in Various Reagents
Doing the coupled assays in detergent solutions should circumvent the aforementioned problems associated with LCP while keeping the advantage of using the convenient Pi-sensor. However, our previous attempts in doing such experiments failed because high levels of phosphate contaminants in the assay mix saturated PBP, the Pi-biosensor. 14 In this work, we revisited this technical problem. First, we needed to identify the sources of Pi contamination. Using the MDCC-PBP, we found that the Pi levels in salts and buffers were negligible, and identified the major source for Pi contamination as being detergents, G3P, and acylP.
Detergents were a necessary component in the assay mix to keep PlsY soluble and were normally kept at three times the critical micelle concentration (CMC). Previously, the protein was solubilized in the DDM that has a CMC of ∼0.01% (w/v). Our tests revealed that the MDCC-PBP (4 μM) became unresponsive to Pi in the presence of 0.03% (w/v) of DDM, suggesting that DDM was an unsuitable solubilizing agent for the PlsY assay. The same experiment also excluded Triton X-100 as the host detergent. On the other hand, the Pi level in LMNG was found to be negligible (∼0.05 mol%). Thus, a 0.003% (w/v) LMNG solution would contain 20 nM Pi, which was stoichiometrically far below the typical concentration of MDCC-PBP (3–5 μM, 111–185 μg mL−1) in the assay mix. Therefore, LMNG was chosen.
The other sources for Pi contamination were the two substrates, G3P and acylP. Previously, we employed an enzymatic method to eliminate Pi by attaching it to a nucleotide through the action of PNPase. 9,14 After the Pi removal, the enzyme was conveniently removed by heating at 65°C followed by centrifugation. This method, while very efficient in treating G3P, could not be directly used for acylP because this mixed anhydride is extremely heat sensitive. 15 The method was then slightly modified. After the Pi-removal reaction, Ni-NTA beads were used to remove His-tagged PNPase instead of heating. However, the removal of Pi was incomplete (from 15 mol% to 0.5–1 mol%). A probable cause was the constant production of Pi as a result of acylP hydrolysis 15 during the lengthy process that includes enzymatic reaction (1–2 h) and Ni-NTA binding (1–2 h).
Next, we sought to remove Pi from the acylP using LCP (Fig. 2). AcylP containing contaminant Pi was reconstituted into LCP at 5 mol% concentration. Upon reconstitution, acylP inserts into the LCP bilayer via its palmitoyl chain and Pi stays in the water channels. LCP was then soaked with Pi-free buffer. Being porous, LCP should release Pi into the bulk soaking solution, in an expected time frame of 2–5 min, based on published data with the cobalt ion. 22 AcylP, on the other hand, was expected to be retained in the lipid bilayer of LCP. Consistent with this, no residual Pi could be detected using MDCC-PBP after soaking three times. The LCP, now composed of ∼57% (w/v) of monoolein, ∼3% (w/v) of acylP, and ∼40% (w/v) of water, was dissolved in DMSO to generate a 30 mM acylP stock solution. The stock was stable for at least 6 months at −80°C.
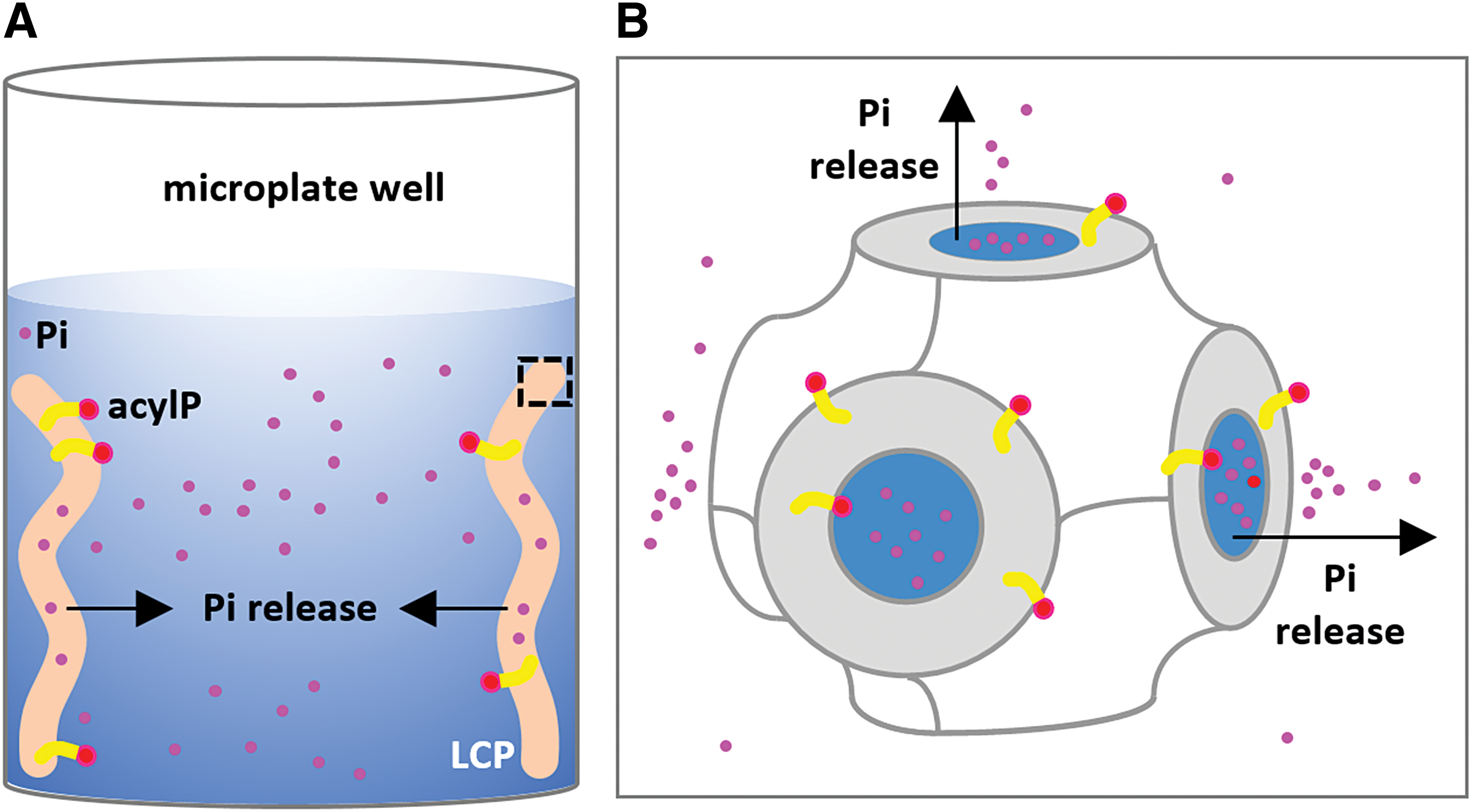
Schematic drawing of the selective removal of Pi from acylP using LCP. LCP reconstituted with acylP is soaked with Pi-free buffer
Assay Establishment and Optimization of Enzyme Loading
Now that the background Pi was reduced to a negligible level, the nascent Pi generated by PlsY reaction could be monitored conveniently. As shown in Figure 3A, the fluorescence of MDCC-PBP increased with time in an enzyme- and substrate-dependent manner. The enzyme concentration used was 16 ng mL−1, and the G3P and acylP concentrations were 10 mM and 20 μM, respectively. Under these conditions, the zero-order region of the progress curve was very narrow (∼3 min).

Activity assay of PlsY in detergents.
To seek conditions that allow a longer linear region, activity assays were performed under a series of PlsY concentrations. As shown in Figure 1B, the linear region became longer at lower enzyme concentrations. At 0.63 ng mL−1, the reaction velocity was constant up to 30 min. This experiment also revealed that the PlsY activity increased linearly at the enzyme concentration range of 0–5 ng mL−1; at higher concentrations, the reaction rate started to level off (Fig. 3C). Accordingly, the enzyme concentration was always kept between 0–5 ng mL−1 thereafter.
Determination of Kinetic Constants
High-throughput screening for competitive inhibitors of enzymes is usually performed under substrate concentrations around or below K m. Therefore, we next set to determine the K m for both substrates. PlsY activity assays were performed by varying one substrate concentration in the presence of saturating concentration of the second substrate. PlsY displayed classic Michaelis–Menten kinetics (Fig. 4), with V max of 57.5 μmol min−1 mg−1 (U mg−1), and K m of 1.14 mM G3P and 6.2 μM acylP.
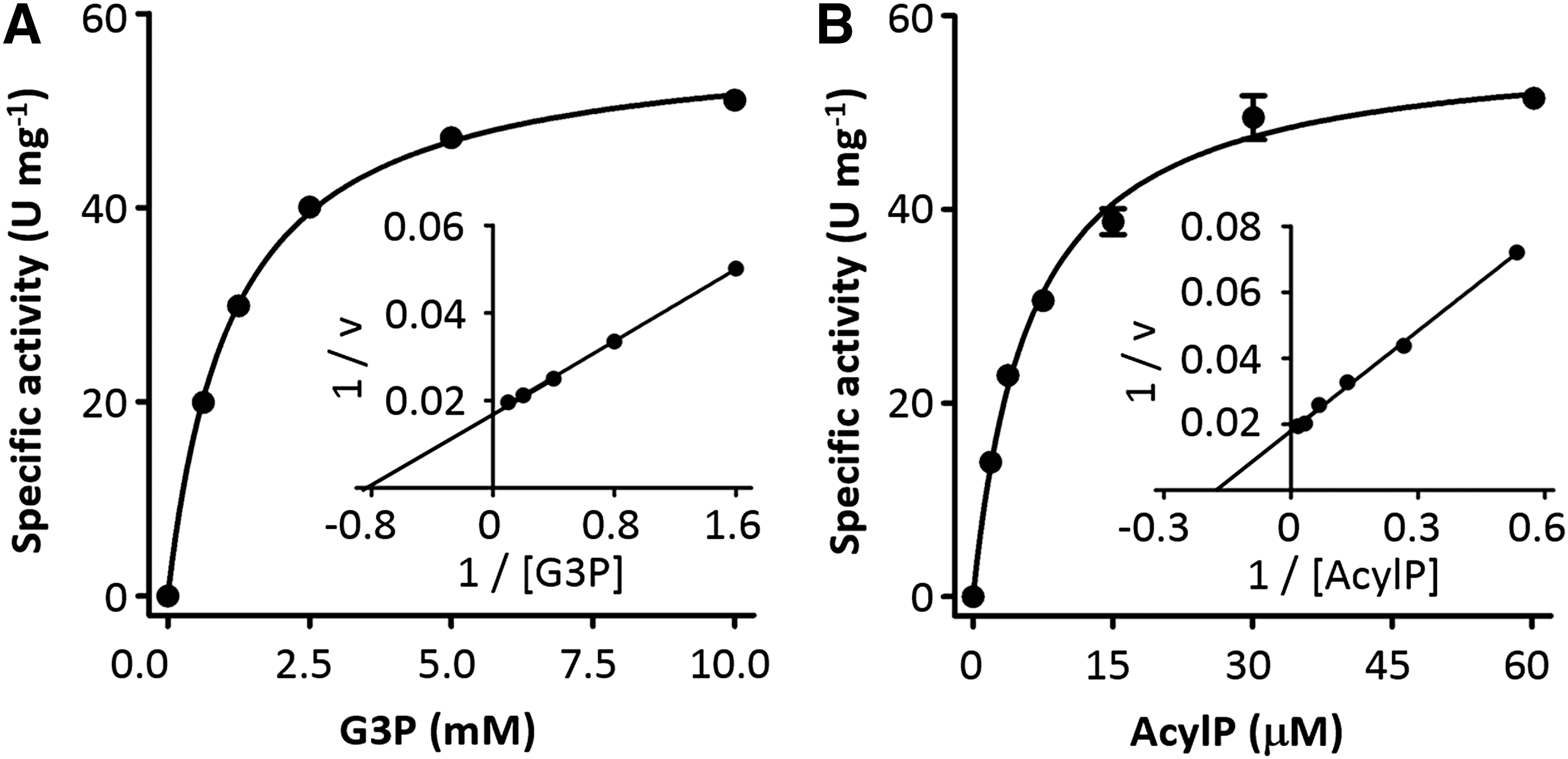
Kinetic parameters of PlsY. The Michaelis–Menten fitting (main panel) and the Lineweaver-Burk plot (inset) were used to determine and to visualize the kinetic constants, respectively. One unit (U) of PlsY is defined as the enzymatic conversion of 1 μmol of the substrate to the product per minute.
It should be noted that the G3P we used contained approximately equivalent amounts of α- and β-isomers, according to the manufacturer. Further, the α-isomer is a mixture of sn-1- and sn-3- stereoisomers, presumably also at equivalent levels. Because PlsY only uses the sn-glycero-3-phosphate isomer, the effective substrate concentration was approximately quartered. This translated the K m value for G3P from 1.14 mM to 0.29 mM, which was comparable to the reported value (0.1 mM) obtained from assays carried out in native membranes. 16
The K m of acylP in our assay was fivefold less than the reported value (30 μM). 16 This might reflect the different diffusion rates of the lipidic substrate in different environments. The carbon chains of LMNG (12 carbons) are shorter than lipids (16–18 carbons); thus, micelles probably have more dynamic structures for more rapid diffusion of acylP. In addition, the micelles are generally 1–2 orders magnitude smaller (in size) than microsomes and thus have a larger surface-area-to-volume ratio. This could mean higher exchange rates of acylP between micelles than between microsomal membranes. It is also possible that the difference in K m values was intrinsic because the PlsY proteins in the two studies were from different organisms; the A. aeolicus PlsY was used in the current study, and the Streptococcus pneumoniae PlsY was used in literature. 16
As a proof-of-principle test for inhibitor screening, it was necessary to characterize a known inhibitor using the current assay system. For this purpose, we chose the product lysoPA, since it was the only biochemically confirmed inhibitor (Fig. 5A). 16 PlsY activity was measured under K m values of G3P/acylP, with various concentrations of lysoPA, in 96-well half-area plates. As shown in Figure 5, lysoPA inhibited PlsY activity in a dose-dependent manner, with the IC50 of 19 μM. This was in good agreement with the reported value of 12 μM from a similar assay using isolated membranes that contained recombinant S. pneumoniae PlsY. 16
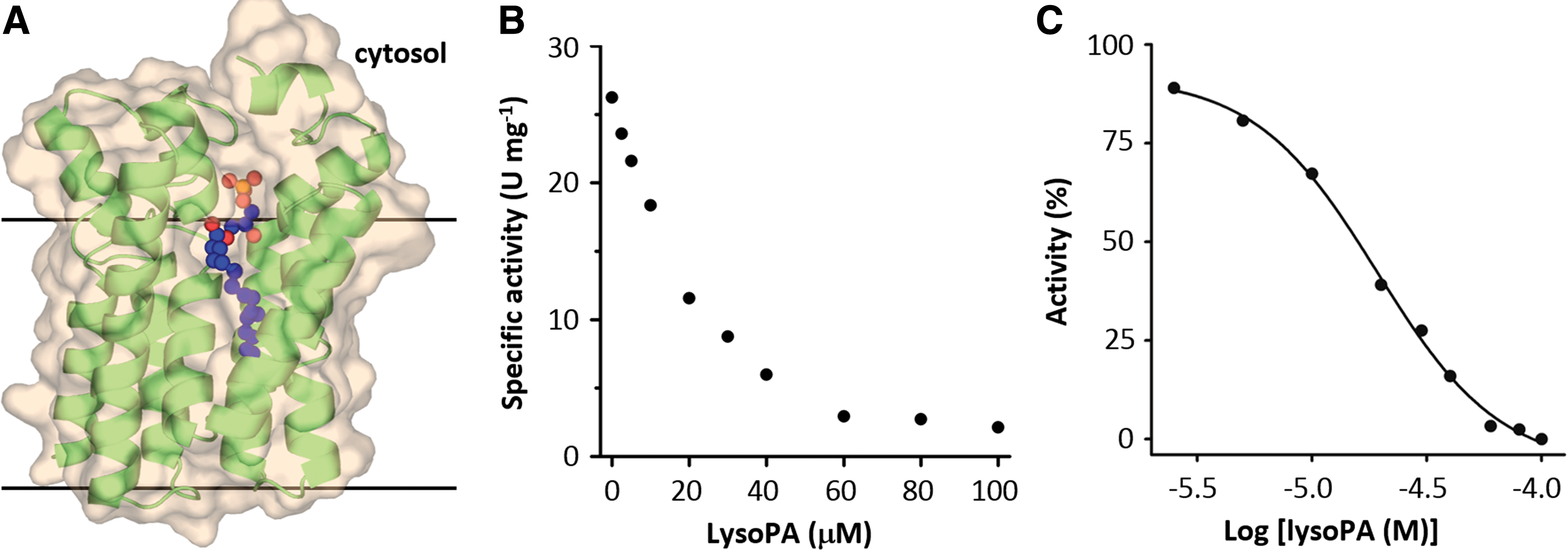
Inhibition of PlsY activity by its product lysoPA.
Thus far, the assays were carried out in a continuous mode for 0.5–1 h, during which time the plate reader was occupied. This can impede the throughput. To run the assay in larger batches (several hundred in parallel), the end-point assay mode is preferred. Under this mode, the throughput depends on the length of the linear region of the assay. As shown in Figure 3B, the velocity was constant in the first 30 min. This window of time should allow the screening of several plates in parallel.
Conclusions
In summary, we have developed a fluorometric assay for PlsY in 96-well format and in a liquid-handling friendly formula. The conditions of the assay were optimized for enzyme loading to ensure long linearity of the progress curve. The kinetic constants were measured using the coupled assay, revealing the Michaelis–Menten relation between the enzymatic activity and the substrate concentrations. In addition, the IC50 of the lysoPA was measured using the assay, providing quantitative information for the product inhibition. The results should encourage further validation using robotics for automated screening of small-molecule inhibitors from chemical libraries against this potential antibiotic drug target.
Authors' Contributions
Y.T.: designed and performed experiments, analyzed results, and assisted article preparation. D.L.: designed the research, analyzed results, and wrote the article.
Footnotes
Acknowledgments
The authors thank Mr. Yitian Luo for the redrawing of the LCP cartoon, and the staff members of the Large-scale Protein Preparation System at the National Facility for Protein Science in Shanghai, Zhangjiang Lab for equipment maintenance and management. This research has been supported by the National Natural Science Foundation of China (31570748, U1632127, and 31870726), Key Program of CAS Frontier Science (QYZDB-SSW-SMC037), CAS Facility-based Open Research Program, and the 1000 Young Talent Program.
Disclosure Statement
No competing financial interests exist.