
Abstract
Multidrug resistance (MDR) is a major health issue for the treatment of infectious diseases throughout the world. Staphylococcus aureus (S. aureus) is a Gram-positive bacteria, responsible for various local and systemic infections in humans. The continuous and abrupt use of antibiotics against bacteria such as S. aureus results in the development of resistant strains. Presently, mupirocin (MUP) is the drug of choice against S. aureus and MDR (methicillin-resistant). However, S. aureus has acquired resistance against MUP as well due to isoleucyl-tRNA synthetase (IleS) mutation at sites 588 and 631. Thus, the aim of the present study was to discover novel bioactives against MUP-resistant S. aureus using in silico drug repurposing approaches. In silico drug repurposing techniques were used to obtain suitable bioactive lead molecules such as buclizine, tasosartan, emetine, medrysone, and so on. These lead molecules might be able to resolve this issue. These leads were obtained through molecular docking simulation based virtual screening, which could be promising for the treatment of MUP-resistant S. aureus. The findings of the present work need to be validated further through in vitro and in vivo studies for their clinical application.
Introduction
Staphylococcus aureus is a coccoid Gram-positive bacterium that grows aerobically or facultatively anaerobically to form yellow golden colonies at 18–40°C. Humans serve as a reservoir for this bacterium, and it is thought that approximately 50–60% of individuals are colonized. More than 15% of them persistently carry S. aureus , which is responsible for causing infections. 1,2 It is a leading cause of superficial skin infections, bacteremia, soft-tissue conditions such as impetigo, scalded skin syndrome, folliculitis, and so on. In addition, it is also involved in pneumonia, bone and joint infections, gastroenteritis, and urinary tract infections. 3,4 S. aureus significantly dominates the rate of bacterial infections around the world because of its ability to develop diverse mechanisms of drug resistance. It is one of the prime culprits for infection in the United States and other industrialized countries. According to a report from the United States, S. aureus was found to be most frequently recovered bacteria from inpatients. Similarly, in Europe, it was observed as the second most recovered bacteria from bacteremia. 5 –7
Penicillin was the first antibiotic used to treat S. aureus infections, but after a long period of use, the bacteria developed resistance due to penicillinase. 8 The continuous quest for an antibiotic active against S. aureus led to the discovery of methicillin. But the appearance of methicillin-resistant strains of S. aureus (MRSA) made the drug clinically ineffective. 9 –13 In this context, mupirocin (MUP) is an important antimicrobial agent produced by Pseudomonas fluorescens. It is used to treat topical infections and nasal decolonizations by inhibiting MRSA. 14 –16
Aminoacyl-tRNA synthetase (AaRS) is a pathogenic enzyme, having metabolic functional role, and it is an established drug target for various antibiotics. 17 AaRS enzymes are multidomain proteins, having a catalytic and anticodon binding domain. They can be divided into two classes, each specific for particular amino acids. The different classes of AaRS perform the same biochemical functions. The reaction take place in two steps. In the first step, acetylation of glutamate (Glu) takes place by covalently binding of one ATP molecule and Glu to the synthetase active site, resulting in the formation of enzyme bound aminoacyl-adenylate and pyrophosphate. In second step, aminoacylated tRNA is formed by amino acid transfer to the tRNA acceptor stem. 18 IleS is one type of AaRS that is present in S. aureus through which MUP shows its antimicrobial activity. IleS belongs to the class I tRNA synthetases, which are characterized by an ATP-binding Rossman fold containing conserved HMGH and KMSKS motifs. Its mechanism of action involves interfering with the bacterial protein synthesis through irreversible and competitive inhibition of IleS. The inhibition of IleS leads to the accumulation of uncharged isoleucyl-tRNA responsible for the synthesis of alarmone guanosine pentaphosphate. The stringent response of alarmone curtails RNA synthesis. 19
The widespread use of MUP in clinical practice and in over the counter (OTC) drugs has led to a resistant strain, in particular MRSA. 20 –22 For S. aureus, three levels of susceptibility have been defined: (1) a minimum inhibitory concentration (MIC) of ≤4 μg/mL, (2) low-level resistance at a MIC of 8–64 μg/mL, and (iii) high-level resistance at a MIC of ≥512 μg/mL. 23 The strain of S. aureus with a MIC >8 μg/mL has shown a point mutation in the region of the Rossman fold adjacent to residues 588 and 631 by changing valine to phenylalanine (Val-to-Phe). This was confirmed further by an allele-specific PCR survey of 32 different strains. 24
Drug repurposing is an emerging field for identifying new biological roles of approved drugs. Repurposing is a cost-effective approach for quickly establishing alternative therapies for existing diseases. 25,26 Drug repurposing methods are applied by using various in silico techniques such as virtual screening of libraries of approved drugs, as well as molecular docking simulation of a drug against new targets. Computer-assisted in silico approaches are used to analyze, integrate, and apply large-scale information retrieved from scientific reports. Repositioning approaches may arise serendipitously or following close clinical observation of existing therapies. Computational drug repositioning provides a systematic and rational solution for recognizing treatment options compared to conventional drugs. 27 –29
The present study aimed to highlight the mutated residues of IleS that are responsible for the development of MUP resistance in S. aureus. Moreover, drug repurposing approaches were applied by virtual screening of the ligand library containing more than 4,500 drugs approved by the Food and Drug Administration (FDA) for developing novel therapies for MRSA.
Materials and Methods
All computational studies were performed using a HP Z240 workstation with 32GB RAM, 2TB storage capacity, and a 2GB NVIDIA graphic card, and with Microsoft Windows v10 as the operating system. The binding interaction of the drug molecules with IleS was performed through molecular docking simulation using the procedure described below.
Selection and Preparation of Protein
A 3D structure model of IleS of S. aureus bound with the MUP ligand (pdb id-1FFY) was obtained from the RCSB protein data bank. 30,31 The MUP ligand bound in the receptor's active binding site was separated from the complex molecule using software chimera (Resource for Biocomputing, Visualization, and Informatics). The receptor molecule was prepared for molecular docking simulation by adding polar hydrogen, removing redundant water molecules and adding Gastgeiger charges with equal distribution among the residues. IleS was bound to tRNA and remained so throughout the studies. The tRNA–IleS complex results in conformational changes in the macromolecular target, thus affecting the binding pattern of the ligand molecule.
Preparation of the Ligand for Molecular Docking
The MUP ligand was prepared for molecular docking simulation by providing the number of rotatable, non-rotatable, and un-rotatable bonds using the AutoDock software (The Scripps Research Institute). 32
Molecular Docking Simulations
The ligand binding site was identified in the IleS enzyme by exploring its binding interactions using Discovery Studio (Accelrys Inc., Biovia). The binding site of the IleS enzyme was utilized to enumerate the grid parameter points of the grid box required to perform the molecular docking simulation of ligand molecule. These grid parameters were utilized for all docking runs. The grid box was placed in the center of the ligand by covering all the binding residues involved in the binding of the ligand to ensure that all the extended conformations of ligand fit within the grid box.
The Lamarckian genetic algorithm (LGA) is the primary conformational search approach used in AutoDock for molecular docking simulation. A trial population was created for various possible conformations, and these conformations were mutated, leading to an exchange of conformational parameters. This is analogous to competitive biological evolution over successive generations, which ultimately selects for structures with the lowest binding energy.
The individual conformational search for local conformational space, finding local minima, and then passing this information on to later generations was performed in a “Lamarckian” fashion, which was an additional feature. The free energy of binding of small molecules to macromolecular targets was predicted by using a semi-empirical free energy force field. This allowed the incorporation of intramolecular energies into the predicted free energy of binding by evaluating energies for both the bound and unbound states based on a comprehensive thermodynamic model. Docking parameter files for each ligand were prepared using the 150 Genetic Algorithm (GA) runs, 250,000 maximum numbers of evaluations, 27,000 maximum numbers of generations, and 0.02% rates of gene mutation. All these docking parameters were saved to a file known as the docking parameter file. 32,33
Docking Method Validation
The positions and orientations of the ligand obtained after the molecular docking simulation represented potential binding modes of the inhibitor molecules. The various docking parameters considered in the docking methods were validated by re-docking individually crystallized ligand MUP over the IleS enzyme. 33 The following parameters were used to validate the molecular docking process for docking IleS with the MUP ligand.
Overlay method
The molecular docking simulation method was validated when the docked conformation of the ligand was perfectly overlaid with the crystal structure of the ligand present in the downloaded protein.
Chemical resemblance
This method was validated when the docked ligand had the same interaction with the residues of the macromolecule as present in the downloaded crystallized macromolecule.
Mutations in the Target Enzyme
Antonio et al. have already reported that the IleS enzyme has developed resistance against the currently existing drug MUP by two point mutations by Val-to-Phe at sites 588 and 631. 34 The above mutations were carried out in the original non-mutated enzyme through Swiss protein data bank viewer (SPDBV) software followed by energy minimization process by using Assisted Model Building and Energy Refinement (AMBER94) force field in GROMACS software. 35,36
Validation of the Mutated Target Enzyme
The mutated target enzyme was validated for its binding affinity against MUP by performing molecular docking simulation and compared it to the non-mutated enzyme.
Virtual Screening of the Mutated Target
The mutated IleS enzyme was screened virtually with a ligand library containing 4,500 FDA-approved drugs obtained from the ZINC database for exploring potential lead molecules. These leads were expected to be potential inhibitors of the mutated IleS enzyme. 37 The detailed schematics of drug repurposing in MUP-resistant S. aureus are presented in Figure 1.
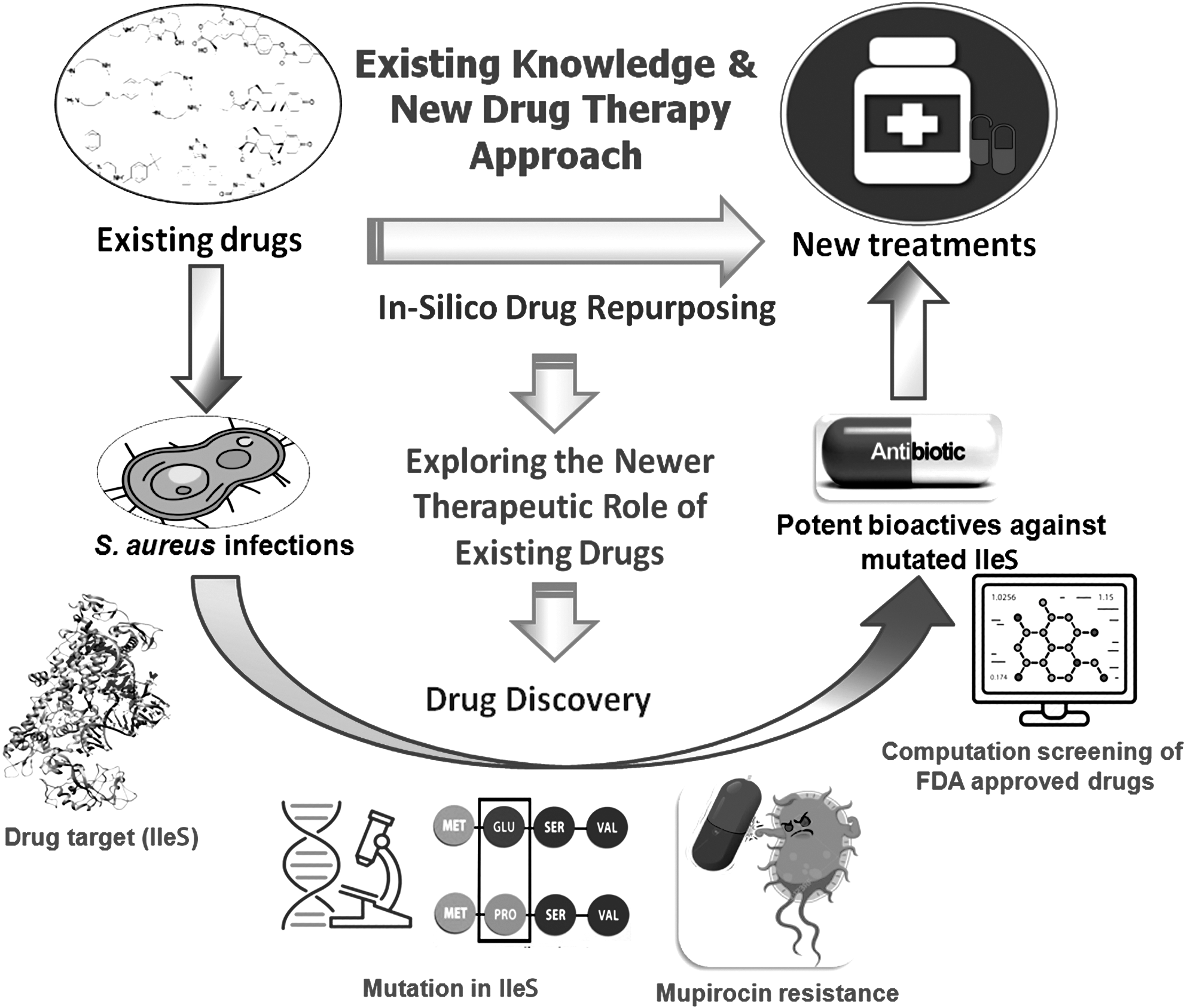
The concept of in silico drug repurposing approach for finding new leads from existing drugs against mupirocin (MUP)-resistant Staphylococcus aureus.
Results
Selection and Preparation of Protein
The 1FFY protein complex of IleS of S. aureus obtained from the RCSB protein data bank consists of a single polypeptide chain of 917 amino acids. The receptor molecule was saved in *.pdbqt format after being processed through the AutoDock software. The detailed structure is presented in Figure 2.
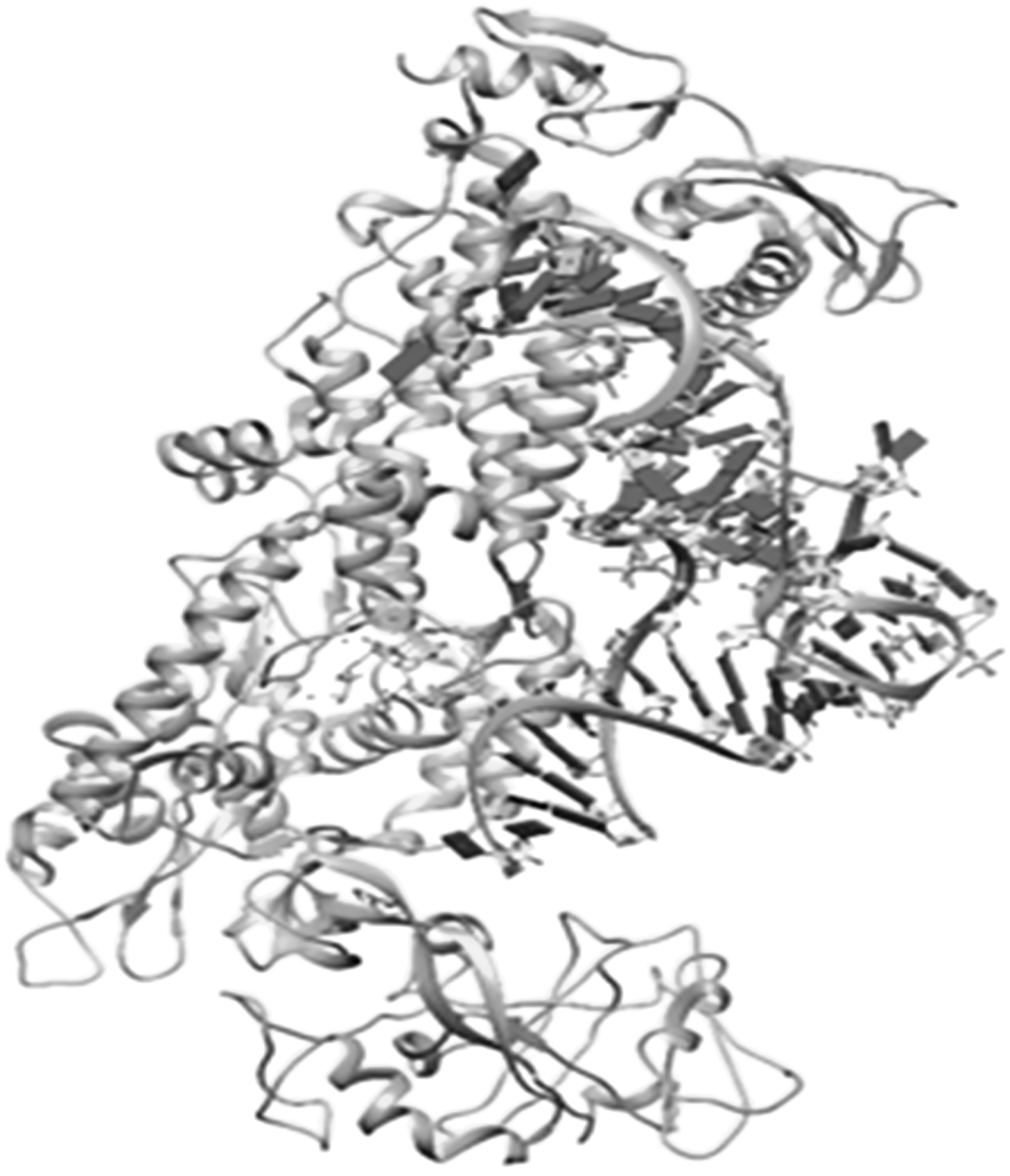
Crystal structure of the IleS of S. aureus. The 3D structure of IleS of S. aureus was obtained from the RCSB protein data bank (PDB ID-1FFY) and processed for molecular docking simulation by adding polar hydrogens and Gastgeiger charges and removing redundant water molecules using the AutoDock software.
Preparation of the Ligand for Molecular Docking
Out of a total of 17 bonds, six rotatable bonds were present in the ligand molecule. These were kept rotatable in the current experimental study. The MUP ligand is shown in Figure 3. The processed ligand molecule was also saved in the *.pdbqt format for performing molecular docking simulation with the AutoDock software.
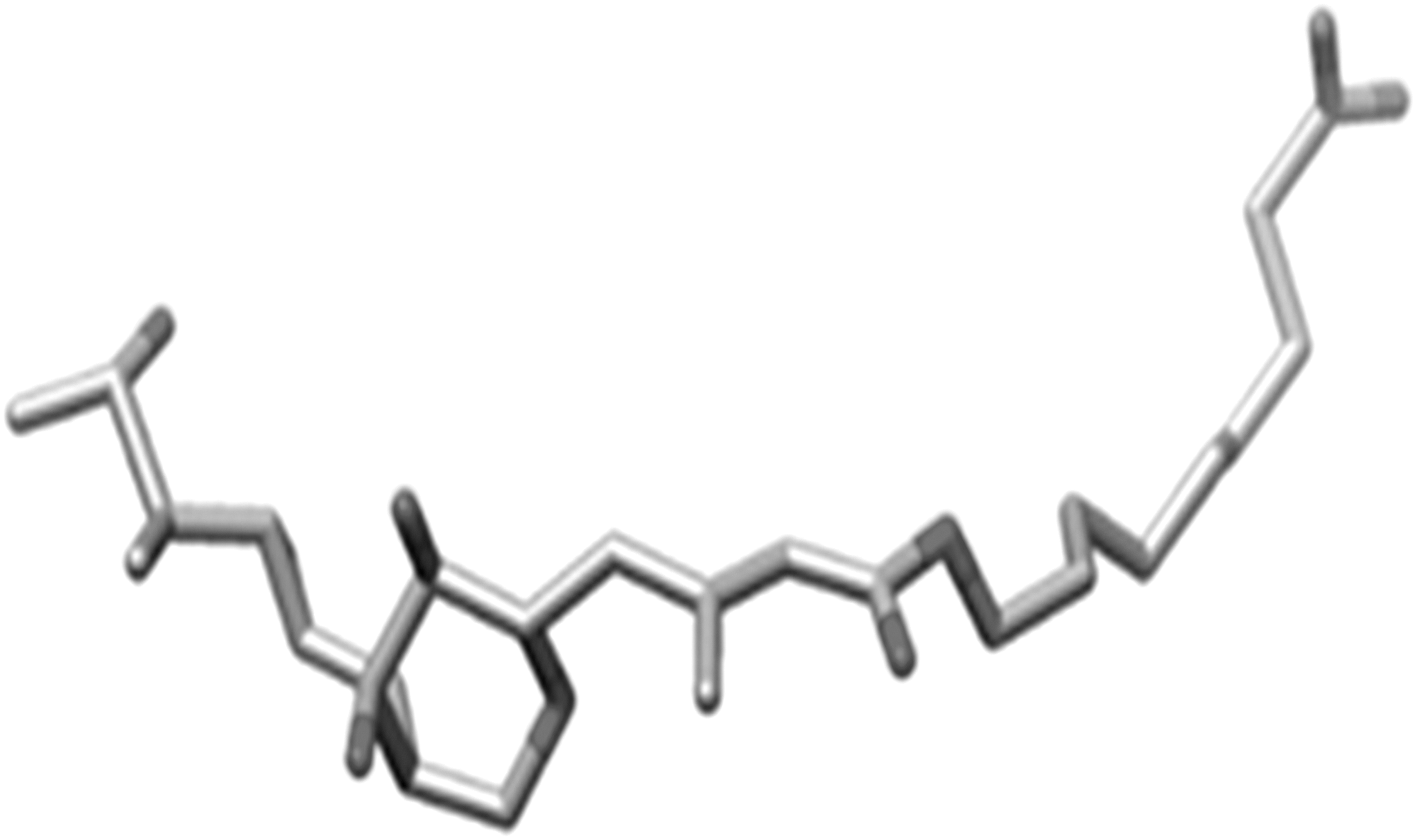
The bound MUP ligand separated from the IleS enzyme. The bound MUP ligand was separated from the macromolecule using the software Chimera.
Identification of the Ligand Binding Site and Grid-Box Formation
The bound MUP ligand interacted significantly with Val588, Gln558, and Gly555 amino acids present in the active binding site of the IleS enzyme. An appropriate grid box was prepared by covering all the macromolecular residues involved in the active binding of the ligand, as shown in Figure 4. The coordinates used to prepare the grid box are given in Table1 .

Three-dimensional grid box for molecular docking simulation of the IleS enzyme. A 3D imaginary grid box covering all the active ligand binding residues present in the receptor molecule was prepared to generate map files using Autogrid software.
Coordinates of Grid Box for the IleS Enzyme
Docking Parameters
The docking parameter file was utilized by the AutoDock software to perform molecular docking simulation of the MUP ligand against the prepared IleS enzyme. The results obtained after molecular docking of the bound MUP ligand with the IleS enzyme are presented in Table 2. The 3D confirmation MUP ligand is shown in Figure 5.

Three-dimensional docked confirmation of MUP. The docked confirmation of MUP in the active binding cavity of IleS enzyme obtained using Pymol software.
Molecular Docking Results of Ligand MUP with the IleS Enzyme (1FFY)
RMSD, root mean square deviation.
Docking Method Validation
The molecular docking simulation of MUP against the IleS enzyme was successfully validated using the parameters detailed below.
Overlay method
The docked conformation of the MUP ligand perfectly overlaid the bioactive conformation of the ligand present in the receptor complex obtained from the RCSB protein data bank, with a root mean square deviation of 0.57. The perfect overlap of the docked conformation of the MUP ligand with respect to its crystallized conformation successfully validates the molecular docking simulation process and the parameters utilized. The overlaid conformation of the docked ligand with reference to the bioactive crystal structure of the ligand obtained from the RSCB protein data bank is shown in Figure 6.

Overlay conformation of the docked ligand. The superimposition of the docked conformation of the ligand with reference to its bioactive conformation of the ligand present in the crystallized structure of the target protein.
Chemical resemblance
The docked MUP ligand has similar binding interactions that were present in the bioactive conformation obtained from the crystallized enzyme complex. The interactions present in the crystal and in the docked structure are shown in Figure 7.

Binding mode and chemical interactions of the bound MUP ligand. The binding interactions of the docked conformation as well as the bioactive conformation were procured using the software DS Visualizer.
Validation of the Mutated Target Enzyme
The poor binding affinity of the MUP ligand against the mutated target enzyme was validated successfully through molecular docking simulation. The result confirmed the development of the resistance by the S. aureus bacteria by two point mutations at sites 588 and 631 by converting Val-to-Phe. The binding energy of MUP against the mutated enzyme is given in Table 3. The schematic representation of the mechanism involved in the development of bacterial drug resistance against MUP is shown in Figure 8. The 2D binding interactions of mutated enzyme are presented in Figure 9.

The mechanism involved in the development of drug resistance by S. aureus against MUP and the schematic approach to develop a newer therapy.
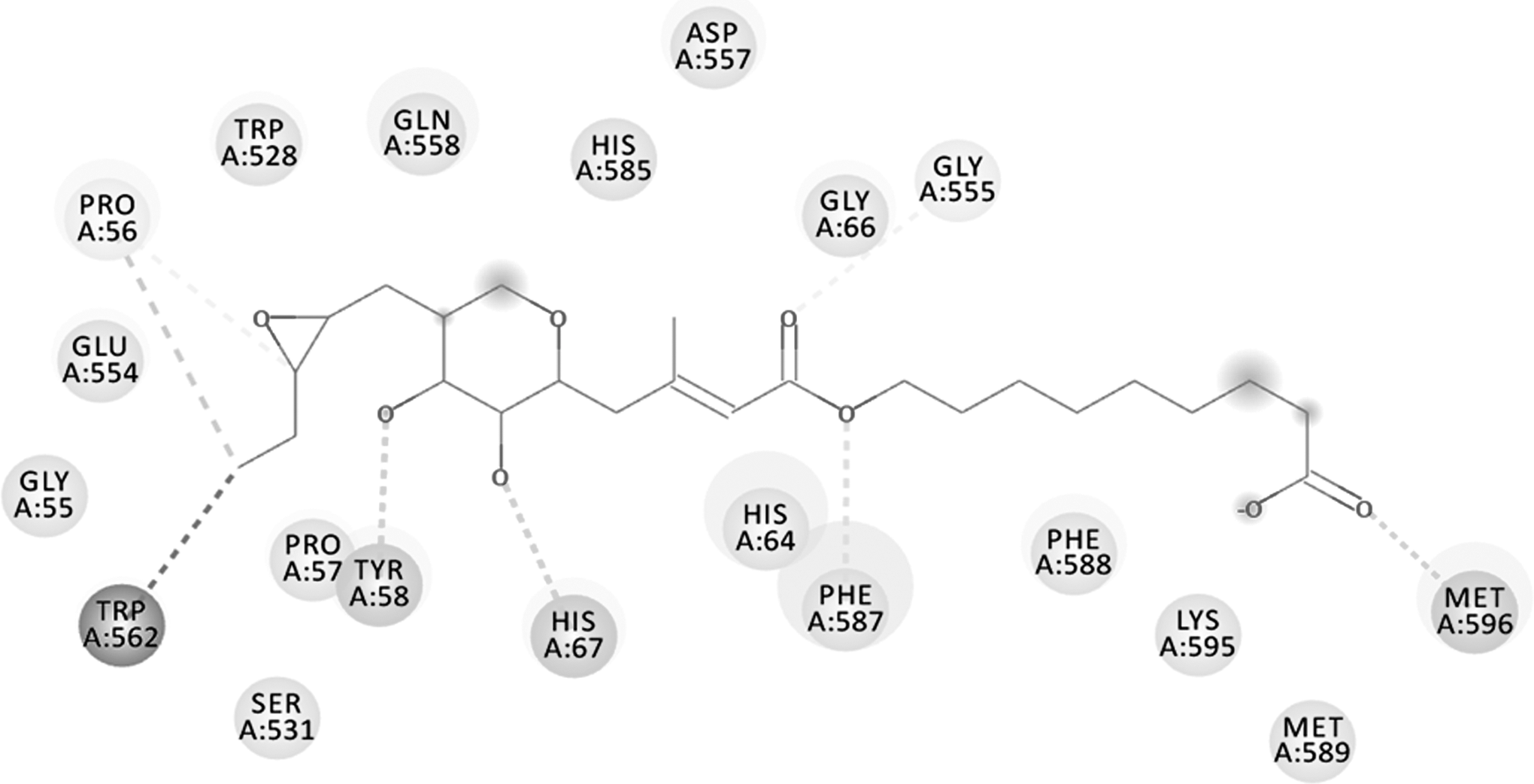
Two-dimensional binding interactions of mutated enzyme. The binding interactions obtained from DS Visualizer clearly describe that the binding pattern of MUP is highly affected because of the two point mutations in the binding residues of the IleS enzyme at sites 588 and 631 by converting valine to phenylalanine, resulting in the development of drug resistance to MUP by S. aureus.
Molecular Docking Result of MUP Against the Mutated IleS Enzyme of Staphylococcus aureus
Virtual Screening of the Mutated Target
Out of ligand library containing 4,500 FDA-approved drugs, drug molecules having potential binding affinity for the mutated IleS enzyme of S. aureus were selected on the basis of the lowest binding energy in the predefined range of −5 to −15 kcal/mol. The binding energy obtained for the top 10 drug molecules is given in Table 4. The 2D and 3D interaction of the top 3 lead molecules (i.e., plerixafor, paliperidone, and buclizine) is shown in Figures 10–12, respectively and the interactions of the remaining ligands are shown in Supplementary Figures S1, S2, s3, s4, 5, 6, 7.
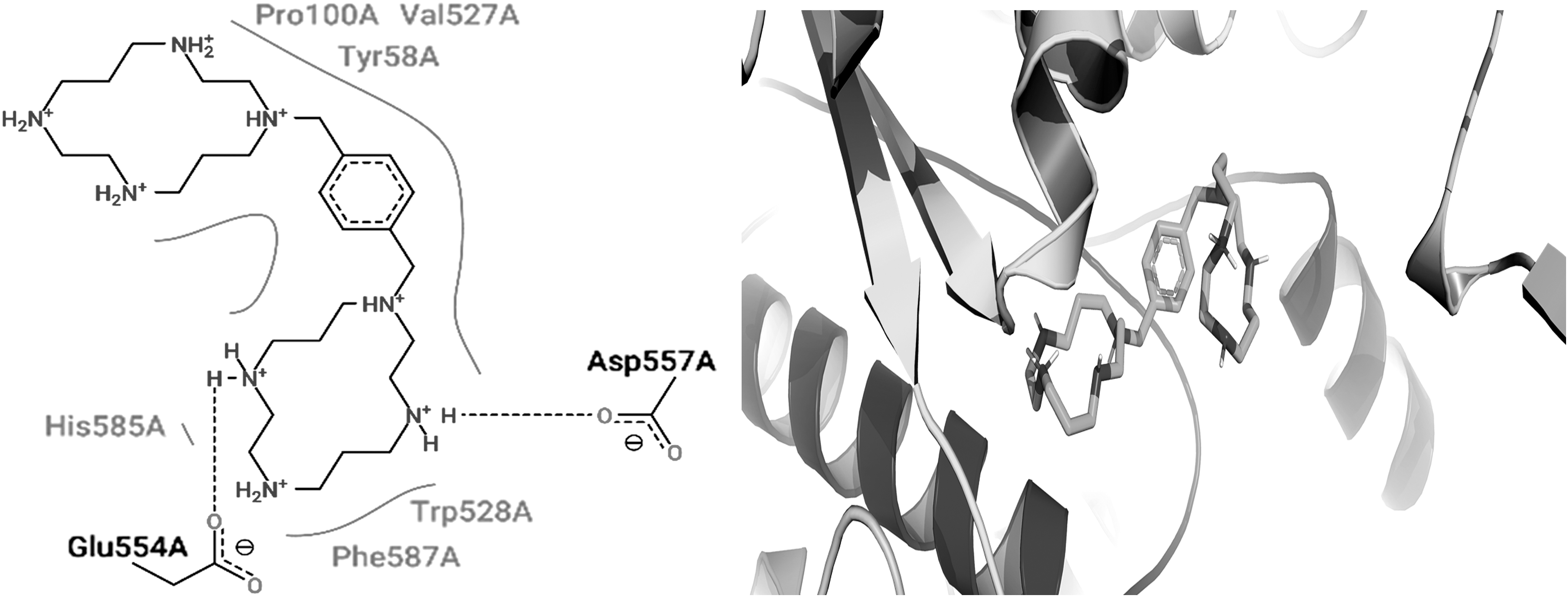
Two- and three-dimensional binding interactions of plerixafor. The binding interactions of plerixafor with the mutated bacterial IleS.
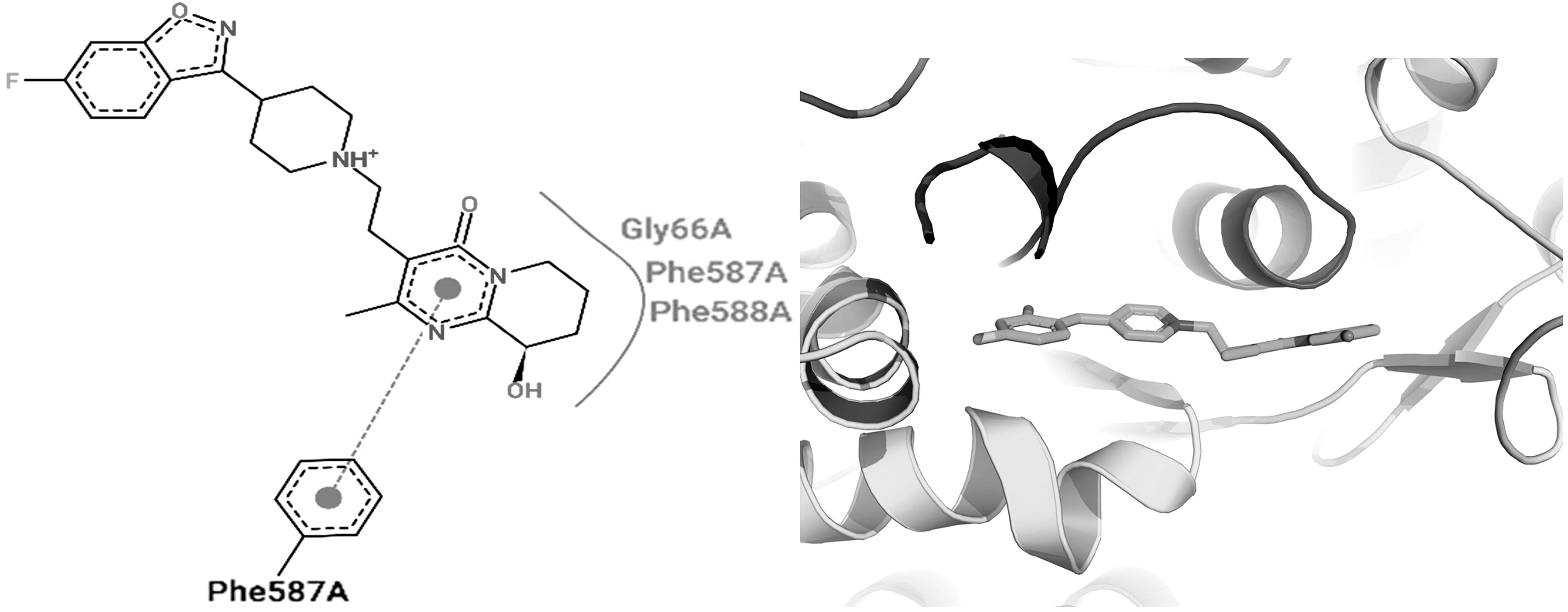
Two- and three-dimensional binding interactions of paliperidone. The binding interactions of paliperidone with the mutated bacterial IleS.

Two- and three-dimensional binding interactions of buclizine. The binding interactions of buclizine with the mutated bacterial IleS.
Binding Energy of the Top 10 Drug Molecules Obtained After AutoDock-Based Virtual Screening of Ligand Library Containing 4,500 Drugs Approved by the Food and Drug Adminstration
Discussion
The random and excessive use of antibiotics for the treatment of bacterial infections has led to MDR development in micro-organisms, especially bacteria such as S. aureus. The bacteria develops drug resistance by causing alteration in the drug targets, enzymatic inactivation of the drugs, increased efflux or reduced uptake, and generation of the biofilms. The development of drug resistance causes remarkable reduction in efficacy of the therapeutic agents, resulting in costlier therapy and problematic patient recovery. The dreadful nature of S. aureus as well as the development of resistance by pathogen against existing therapy is a major problem for pharmaceutical industries worldwide. The problems associated with the development of drug resistance by the pathogen against the existing therapy are countered by continuously developing newer antimicrobial drugs. Discovery of some selective and diverse pathogenic drug targets are highly beneficial for new drug development. The screening of clinically approved drugs through the drug repurposing approach would be highly valuable for developing novel therapies against the diseases affecting humankind.
Drug discovery and development is a very complex process, taking about 15–20 years and costing significant amounts of money. In this context, in silico drug repurposing and repositioning techniques would be promising alternative methods for drug discovery. The drug repurposing approach is economical and time-saving, and is a virtual method of drug screening with a higher success rate. Drug repurposing is a technique in which a newer pharmacological application of an existing drug is identified and established through computational and clinical investigations. In the modern era, these techniques have been widely explored for the successful development of newer therapy for diseases via existing drugs. Previously, newer pharmacological roles for existing drugs were identified either by accident or by observed their clinical manifestations. Nowadays, fast, economical, reliable, and versatile computational repurposing techniques can be utilized to recognize different therapeutic uses for existing drugs.
The development of drug resistance against existing therapy is one of the biggest challenges faced by pharmaceutical scientists working in the field of drug discovery. Pathogenic bacteria such as S. aureus have developed resistance against MUP by causing mutational changes in the target enzyme, IleS. It has been reported that mutational changes at sites 588 and 631 by Val-to-Phe on the IleS enzyme are responsible for the development of drug resistance against MUP.
In the present study, the in silico drug repurposing approach was utilized to develop a novel therapy against the MUP-resistant strain of S. aureus by targeting the mutated IleS enzyme. A 3D structural model of the bacterial IleS enzyme and MUP complex was obtained from the RCSB protein data bank. The bound drug, MUP, was separated from the enzyme complex with the help of Chimera software. The separated drug molecule was docked in the active binding site of the target protein using AutoDock software to validatie the parameters utilized in the molecular docking process. Further, the target bacterial enzyme was mutated by exchanging the Val-to-Phe at both sites (i.e., 588 and 631). 34 The mutated bacterial enzyme was again docked with the MUP ligand, and poor binding interactions were also observed by molecular docking, confirming the development of drug resistance by S. aureus against MUP.
The findings of the current in silico virtual screening methods could lead to the development of drug molecules such as plerixafor, paliperidone, buclizine, metocurine, paliperidone, tasosartan, irinotecan, testosterone, emetine, and medrysone against the mutated target enzyme (IleS) of S. aureus.
Plerixafor is a water-soluble drug that antagonizes chemokine receptor type 4 for treating non-Hodgkin lymphoma and multiple myeloma. Plerixafor binds in the active binding cavity of bacterial IleS mainly by interacting with the Glu554, Asp557, and Phe587 residues. Paliperidone is an active metabolite of risperidone that antagonizes dopamine D2 and the serotonin 5HT2 receptor used for the treatment of schizophrenia. Phe587 and Gly66 of bacterial IleS were found to interact with paliperidone. Buclizine is rapidly absorbed and metabolized after oral administration and antagonizes H1 histaminic receptor for antiemetic, CNS depressant, and anti-vertigo action. Buclizine was found to interact with the Phe587 and His67 residues of bacterial IleS. Tasosartan is an orally active and long-acting angiotensin II blocker used for the treatment of hypertension. Tasosartan strongly interacted with the Phe587, Gly66, and His67 residues of bacterial IleS. Irinotecan is an antineoplastic agent used for the treatments of colorectal and pancreatic neoplasms by interfering with nucleic acid synthesis at the S-phase by inhibiting DNA topoisomerase-I. The binding residues Phe587, Gln558, and Gly555 of bacterial IleS enzyme were found to interact with irinotecan. Emetine exerts antiemetic and anthelmintic activity by interfering DNA replication early in the S-phase in eukaryotic cells. Emetine interacted with residues such as Asp557, Phe587, and Phe588 of bacterial IleS. Medrysone has anti-inflammatory, anti-allergic, and metabolic actions mediated through agonistic action on the corticosteroid hormone receptor. 38 Medrysone bound with the bacterial IleS enzyme by interacting mainly with the His64 and Glu554 residues. The structure–activity relationship of the shortlisted lead molecules revealed on the basis of the in silico binding pattern with the mutated bacterial IleS enzyme clearly suggests that residues such as Phe587, His64, Phe588, and His67 play an important role in the binding of the drug molecule with the mutated bacterial drug target. The current study found that residue Phe587 of the bacterial IleS enzyme interacted with all the shortlisted lead molecules, and this suggests that this residue is vital for inhibition of the IleS enzyme to enable antibacterial activity.
Repurposing techniques are highly promising for reproducing new biological roles for existing drugs. The identified bioactive molecules may be useful for developing novel therapies against skin diseases caused by MUP-resistant S. aureus in the near future. These drugs should be explored further through preclinical and clinical studies in order to validate the proposed hypothesis.
Conclusion
The in silico drug repurposing technique is a highly effective approach for identifying an existing drug molecule that may have therapeutic activity against the MUP-resistant strain of S. aureus. The schematic representation of the mechanism involved in the development of bacterial drug resistance against MUP is shown in Figure 8. A ligand library containing 4,500 drug molecules approved by the FDA was screened virtually against the mutated IleS enzyme of S. aureus. Plerixafor, paliperidone, buclizine, metocurine, paliperidone, tasosartan, irinotecan, testosterone, emetine, and medrysone were found to be potential lead molecules against the mutated IleS enzyme of S. aureus. The molecules established for neurological as well as anticancer therapy supposedly affect healthy individuals and result in undesirable side effects. Based on their safety profile, buclizine, tasosartan, emetine, and medrysone were selected as safe and effective drug candidates for developing therapy against MUP-resistant S. aureus.
Footnotes
Acknowledgments
The authors are thankful to the IPR, GLA University, Mathura, for providing all the necessary facilities to complete the work.
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7