
Abstract
Naproxen (NAP) is one of the commonly used nonselective Cycloxygenase (COX) inhibitors. It is a choice of drug for anti-inflammatory activity by subsiding the generation of the inflammatory components called prostaglandins. The common problem associated with the NAP is gastrointestinal toxicity. It may cause ulceration or stomach bleeding. In this study, the different derivatives of NAP were designed by using phytophenols with the aim that they exert the antioxidant activity and have the potential to reduce ulcer formation. The lead molecules were designed by molecular docking-based virtual screening against human COX-2 enzyme through AutoDock. Then these derivatives were screened for pharmacokinetic profiling by considering Lipinski's filter. The potent and safe molecule was identified by pharmacokinetics and toxicity evaluation. The potent compound was synthesized in the laboratory, purified, characterized, and its pharmacological activities were evaluated. The resultant compound was found to be equipotent and less toxic than the parent compound.
Introduction
NSAIDs are the agents used against inflammation. They also have analgesic activity. The naproxen (NAP) is used in the current study as a parent drug, which is nonselective in nature. Due to its nonselective nature, it exerts the gastrointestinal (GI) side effects. 1,2 The profound use of NAP leads to gastric ulcers or bleeding in the stomach. 3,4 The problem of GI toxicity is due to inhibition of COX1 enzymes, which is responsible for the protection of gastric mucosa, regulation of gastric acid, and responsible for normal function of the body. 5 –7 The parent drug was modified by conjugating it with phytophenols. The phytophenols were chosen with the aim that they exert antioxidant activity so they will block the mechanism of reactive oxygen species and would show beneficiary activity. The phytophenols selected were eugenol, vanillin (VN), carvacrol, sesamol, umbelliferone, menthol, syringaldehyde, thymol and quercetin. 8,9 The derivatives of selected phytophenols with parent drug docked against Cycloxygenase-2 (COX-2) enzymes. This in silico study was performed with the aim to save the time, money, resources, and establish the mechanism of action of newly designed drug molecules. 10,11 The screened active and most potent compound was further synthesized in the laboratory, characterized, and tested for its pharmacological actions.
Experimental Work
Materials and Methods
Materials
The three-dimensional structure model of the COX-2 enzyme complexed with the ligand mefenamic acid is downloaded from the protein data bank and its molecular docking simulation-based in silico virtual screening was performed by using AutoDock 4.2 software. The melting point apparatus was used to record the melting point. Shimadzu UV-1800 ultraviolet/visible spectrophotometer having quartz cells was used for the experiment. The UV method was developed for a synthesized drug in methanol using 256 nm absorption maxima following linearity in between 10 and 60 μg/mL. Infrared spectrum was scanned using Shimadzu IRAffinity-1 FTIR spectrophotometer, using DRS 8000A accessory technique. The Brucker Aviance II 400 MHz using CDCl3 as the solvent was used to record 1 H NMR. Jeol Sx 102/DA-600 mass spectrometer/Data System using electron ionization technique was used to get the mass spectrum. The thin-layer chromatography using silica gel G is used to check the purity of the synthesized drug. Iodine vapors are used for visualization. Mobile phase used was Methanol:Benzene (2:8). NAP and VN were obtained from YARROW CHEM PRODUCT, Mumbai 400086, India. Analytical/spectroscopic-grade chemicals were used in the whole experiment.
In silico molecular docking simulation
The three-dimensional structural model of human COX-2 enzyme complexed with mefenamic acid was downloaded from the protein databank database. The bound ligand was separated from the complex by using Chimera software. 12 The preparation of receptor and bound ligand mefenamic acid for molecular docking was performed by using AutoDock software. 13 The grid points were enumerated by considering the ligand and all the residues interacting with it to make certain that the ligand's extended conformations fit well in the grid box. The grid parameter file consisting of these grid points was utilized by Autogrid utility of the AutoDock suite for generating various maps for receptor as well as ligand, which were further utilized by AutoDock for performing docking simulations. 14,15
Docking parameter file consisting of various parameters utilized for performing molecular docking of the COX-2 enzyme was prepared by AutoDock. The AutoDock software utilizes Lamarckian genetic algorithm as its primary conformational search algorithm for performing the docking studies. The ligand's probable binding pattern was obtained on the basis of their position and orientations identified after the molecular docking simulations. The parameters included in the current in-silico study were validated by performing docking of the human COX-2 receptor against the crystallized ligand, mefenamic acid. The docking of the COX-2 receptor was validated by considering the binding energy, chemical interaction, and its overlay of docked conformation of the ligand. The parameters used in the docking studies were validated if the binding energy of the bound ligand should be in the predefined empirical range of −5 to −15 kcal/Mol, having similar binding interactions and perfect overlay of the docked conformation with reference to its bioactive conformation. 11,16
The validated docking parameters were further utilized to perform comparative study of various approved COX-2 inhibitors. The comparative docking study is performed to select the standard molecule having on the basis of best binding score as well as potential binding interactions with the residues present in the active cavity of the macromolecular target. Ibuprofen, fluprofen, fenoprofen, ketoprofen, NAP, and suprofen were used in the current comparative analysis for identification of the molecule having most potent binding interaction against the COX-2 enzyme.
The NAP nucleus is substituted with different moieties to prepare some of their esteric derivatives. These molecules were virtually screened against human COX-2 enzyme to identify possible leads. The identified leads are supposed to have potential affinity for the human COX-2 and possessing potential anti-inflammatory activity. The potential leads were shortlisted by considering the minimum binding energy in the predefined empirical range and are further evaluated for their pharmacokinetics and toxic effects by using Data Warrior software. This tool predicts the drug likeness and score for the leads by considering their physicochemical properties. 17,18
Synthesis
The Steglich esterification method was utilized for the synthesis of an NAP derivative (NVN) from NAP and VN. The drug NAP and VN in equimolar (10 mM) concentration were taken in 250 mL flat-bottomed flask having 50 mL of dichloromethane. The reactants were kept at 0°C temperature throughout the reaction. The equimolar amount of N, N′-dimethylaminopyridine (DMAP) and N, N′ dicyclohexylcarbodiimide (DCC) were added in portions slowly. The resulting solution was stirred at room temperature for 4 h. The N, N′ dicyclohexylurea was precipitated during the reaction to get dislodged by filtration. The layer of dichloromethane was washed two times with 0.5 M hydrochloric acid (20 mL) and then with 5% w/v sodium bicarbonate (20 mL). Then furthermore, the organic layer was dried over anhydrous thenardite, filtered, and the solvent was removed under reduced pressure. The final product was obtained as solid and recrystallized with methanol (Fig. 1).
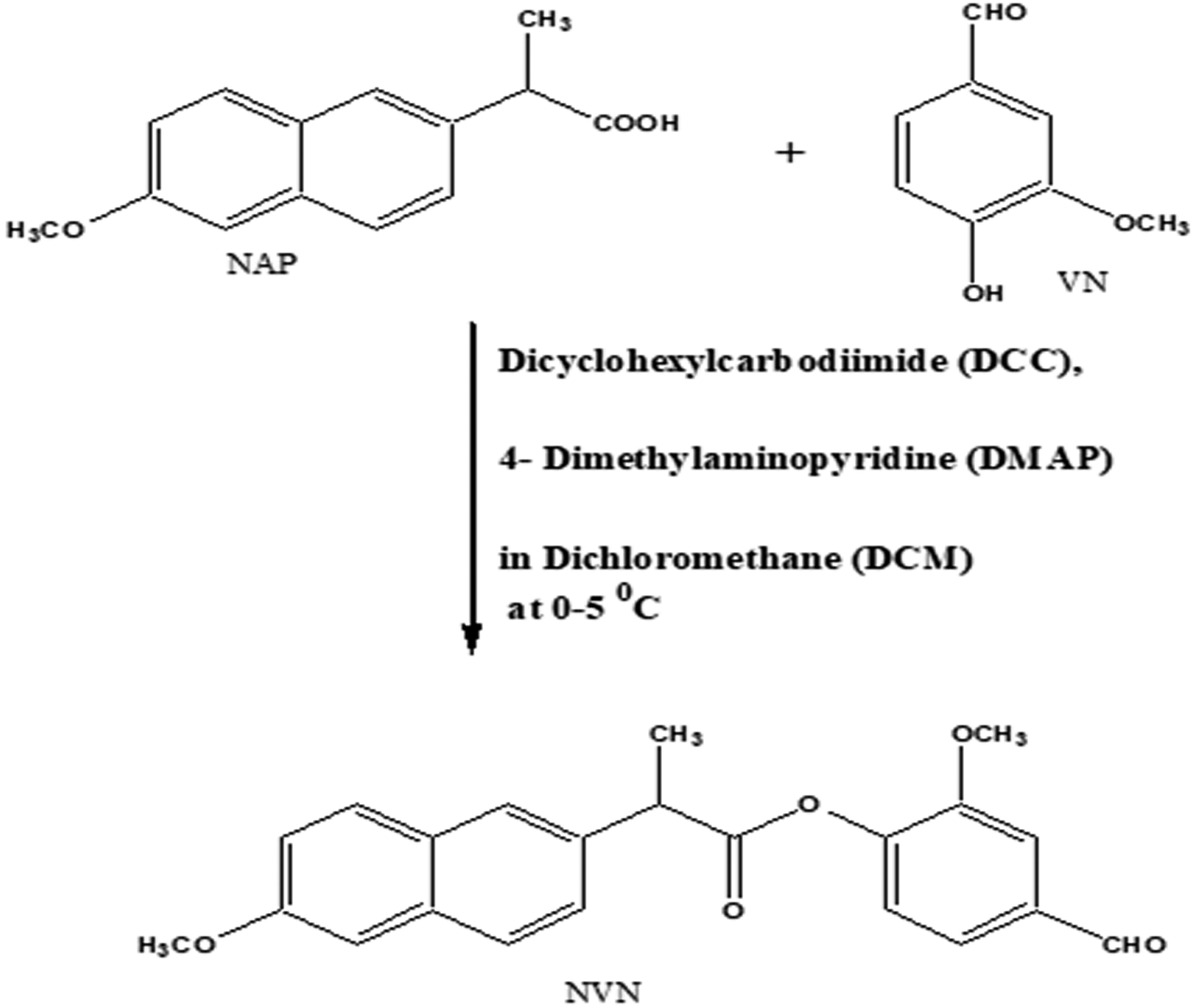
Synthetic scheme.
Characterization of the synthesized drug
Solubility and partition coefficient
Different solvents were used to check the solubility of synthesized drug. n-octanol/phosphate buffer (pH 7.4) was used to determine the partition coefficient of the synthesized drug. The NVN (1 g) was taken in equal volumes of phosphate buffer and n-octanol. Both the solutions were taken in a separating funnel shaken for 2 h at room temperature and left for 1 h. The sample (10 mL) of the phosphate buffer layer was taken and extracted three times with 5 mL of dichloromethane. Thereafter, the respective phases were analyzed by the developed UV assay.
Kinetics of hydrolysis in phosphate and HCl buffer
The kinetics was studied using United States Pharmacopeia (USP) apparatus II, that is, paddle type using isotonic phosphate buffer (pH 7.4) and HCl buffer (pH 2) at 37°C. It is used to determine the rate of chemical hydrolysis. The drug NVN (1 g) was weighed accurately and methanol was used to dissolve it in a 100-mL volumetric flask. The volumetric flask was kept at 37°C ± 1°C for 10 min and the solution was poured to a vessel of dissolution apparatus containing 900 mL of 0.1 M hydrochloric acid. The paddle of apparatus was stirred at 100 rpm and a solution of 10 mL was withdrawn at an interval of 30 min for up to 3 h and replaced with an equal volume of fresh 0.1 M hydrochloric acid immediately. The withdrawn solution was extracted three times with 5 mL chloroform. The chloroform layer was dried further. The residue collected was dissolved in methanol and diluted suitably to estimate the drug. The same procedure was utilized for analysis of drug in the phosphate buffer (pH 7.4).
Pharmacology
The animal experimental protocols were as per animal ethics guidelines and approved by the University Animal Ethics Committee of GLA University, Institute of Pharmaceutical Research, Mathura, UP, India. (1260/PO/Re/S/09/CPCSEA) dated February 23, 2019. Animals were kept at standard conditions where the temperature was 25°C ± 2°C, fed standard animal feed and water ad libitum, kept in 12-h dark–12-h light cycle along relative humidity of 45%–55%. Synthesized drug, along with NAP, was evaluated for analgesic, anti-inflammatory, and ulcerogenic activity. The synthesized drug was compared with NAP for these activities. The methods used for these pharmacological evaluations were as follows.
Anti-inflammatory activity
The anti-inflammatory activity was judged by the Carrageenan-induced paw edema method. 19 Albino rats of Wistar strains, weighing 150–200 g were used. These animals were fasted overnight before test. The rats of either sex were divided into three groups of six animals each. (1) vehicle (control); (2) NAP (standard, 53 mg/kg b.w., 230 mM, p.o.); and (3) NVN (84 mg/kg b.w., 230 mM, p.o.). The suspension of the drug and standard drug was made using carboxymethylcellulose (0.5%, CMC) and given orally. The Control animals were given the 0.5%, CMC.
Analgesic activity
Abdominal writhing assay method was used to determine the analgesic activity.
20
The swiss albino mice, weighing 20–25 g, of either sex were divided into three groups of six animals each. The carboxymethylcellulose (0.5%, CMC) was used to prepare the solutions, the synthesized drug, and NAP were given orally before the freshly prepared acetic acid solution (0.6%, 10 mL/kg) intraperitoneally. The readings were recorded for 20 min as writhes (constriction of the abdomen, turning of trunk, and extension of hind limbs) for each animal. The average number of writhes in each group of drug-treated mice was compared with that of control group and the degree of analgesia was expressed as % inhibition as follows:
where, N t = number of writhing in drug-treated mice
N C = number of writhing in control
Ulcerogenic study
The albino Wistar rats were used for the activity; they were fasted overnight before study. 21 The animals were divided into three groups of six animals each. The doses given were 10 times of anti-inflammatory activity. Animals were treated with NAP and NVN. Animals were sacrificed 12 h after the treatment. The stomach was removed, opened along the greater curvature, washed with saline, and observed for ulcers. The ulcers were scored as: 0 = no observable damage; 1 = superficial ulcers; 2 = deep ulcers; 3 = perforation.
Statistical analysis
Statistical analysis was carried out in vivo studies data. The ulcer index data were subjected to student t-test (unpaired), analysis of variance test, followed by Dunnett's test for determining the levels of significance in antioxidant studies. p Values <0.05 were considered statistically significant.
Results and Discussion
In-silico molecular docking simulation
One out of two identical chains of 551 amino acids present in the macromolecular complex was procured by deleting another one by using Chimera software. The macromolecule was prepared for docking by removing unnecessary water molecules, addition of polar hydrogen, followed by addition and equal distribution of Gastgeiger charge. Maximum possible flexibility is provided to the ligand in the current study by keeping all the available three bonds rotatable. The crystallized structure of human COX-2 enzyme complexed with mefenamic is shown in Figure 2. 16
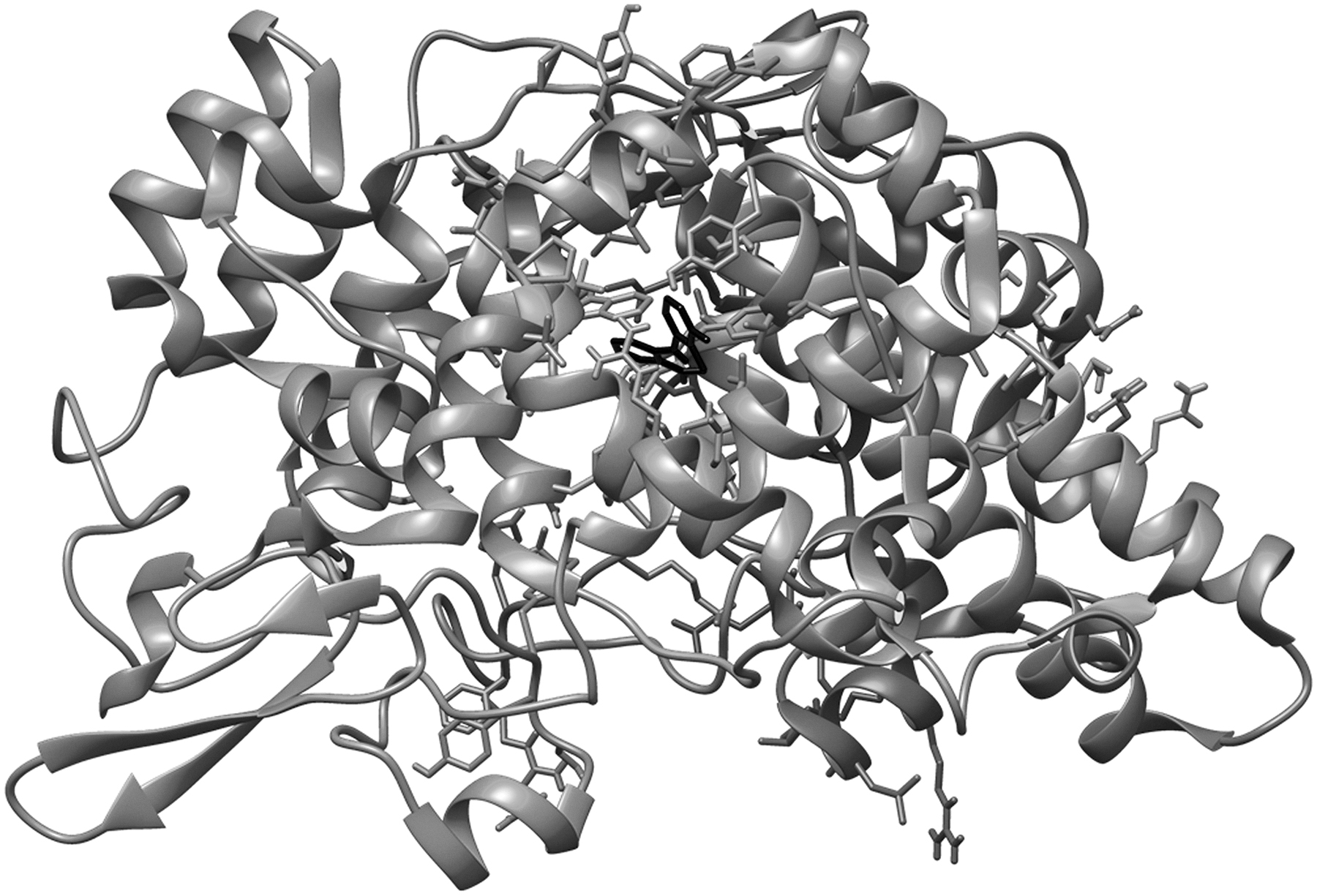
Structure model of the crystallized human COX-2 enzyme complexed with mefenamic acid. COX-2, cycloxygenase-2.
The macromolecular residues Tyr385 and Ser530 were involved in the active interactions with mefenamic acid. The grid coordinates utilized for making three dimensional imaginary grid box are shown in Table 1 and its docking results are tabulated in Table 2.
The Grid-Coordinates of the Grid-Box
Comparative Study of Approved Cycloxygenase-2 Inhibitor Molecules
NAP, naproxen.
The molecular docking process of mefenamic acid against human COX-2 enzyme successfully validated as the binding energy was −7.67 kcal/Mol and lies well within the predefined empirical range. The docked conformation of the ligand was having similar interactions and it was perfectly overlaid with respect to its bioactive conformation. The docked conformation having a perfect overlay with respect to its bioactive conformation is represented in Figure 3.
The superimposed docked conformation of mefenamic acid with respect to its bioactive conformation.
The binding interactions of the docked ligand with reference to its bioactive crystal structure are shown in Figure 4.

Binding mode and chemical interactions of the bound ligand mefenamic acid within the active ligand-binding site of COX-2 receptor of human.
The comparative analysis of the approved COX-2 inhibitors on the basis of the observed binding energy as well as their binding interactions with the macromolecular target clearly signifies NAP as the most potent COX-2 inhibitor. Thus NAP is considered as a standard molecule in the current study and was further utilized for developing a ligand library by generating its various esteric derivatives as COX-2 inhibitors. The result of the comparative study of existing COX-2 inhibitors is shown in Table 2.
The prepared ligand library of the esteric derivatives of NAP were virtually screened to identify potential COX-2 inhibitors. The structure activity relationship obtained after evaluating the binding pattern of all the lead molecules with the binding residues of the human COX-2 enzyme reveals that all the lead molecules are interacting with the Val349, Leu352, and Ala527. Thus, Val349, Leu352, and Ala527 residues of human COX-2 enzyme are found to be key residues playing an important role in the binding of the inhibitor molecule. The other residues, such as Trp387, Phe518, Tyr385, Ser530, Met522, Arg120, Val523, etc. are also having crucial role in the binding interaction of the large proportion of lead molecules. The docking results of the virtually screened ligand molecules against COX-2 enzyme are tabulated in Table 3.
Binding Energy of the Virtually Screened Leads Against Cycloxygenase-2 Enzyme
NAEU, naproxen derivative with eugenol; NCAR, naproxen derivative with carvacrol; NMEN, naproxen derivative with menthol; NQR, naproxen derivative with quercitrin; NSES, naproxen derivative with sesamol; NSYR, naproxen derivative with syringaldehyde; NTHY, naproxen derivative with thymol; NUM, naproxen derivative with umbelliferone; NVN, NAP derivative.
All the designed NAP-based esteric leads were further evaluated for their pharmacokinetics by considering important physicochemical properties. The properties like calculated partition coefficient (cLogP), two-dimensional polar surface area (2D PSA), molecular weight, hydrogen bond donor (HBD), and hydrogen bond acceptor (HBA) sites, etc. were evaluated by using online program Osiris Molecular Property Explorer. The physicochemical properties of the shortlisted lead molecules for human COX-2 enzyme are shown in Table 4.
“Lipinski's rule of five” for the Lead Molecules Targeting Cycloxygenase-2 Enzyme
2D PSA, two-dimensional polar surface area; ClogP, calculated partition coefficient; HBA, hydrogen bond acceptor; HBD, hydrogen bond donor; Mol. Wt., molecular weight.
Later, these leads were evaluated for the presence of any major toxic effect, drug-likeness, and drug score values. It was observed that three out of five selected lead molecules, that is, naproxen derivative with quercitrin (NQR), naproxen derivative with sesamol (NSES), and NVN are having a good pharmacokinetic profile with the presence of no or very low toxic effects. The ADME and toxicity results of best lead molecules obtained after virtual screening are shown in Table 5.
Toxicity Prediction of Proposed Lead Molecules for Cycloxygenase-2 Enzyme
Chemistry
The chemistry behind the scheme was that NAP and the DCC were able to form an O- acylisourea midway, the DMAP served as stronger nucleophile, thus reacted with O-acylisourea, gives a reactive amide (“active ester”). This intermediate could not form intramolecular side products but reacted faster. DMAP acted as an acyl transfer reagent, and subsequent reaction with phenol gives the ester. The mp and percentage yield of NVN was found to be 112°C–114°C and 55.2%, respectively. UV (λ max) nm: MeOH: 231, 256, IR (KBr), cm−1: 3,057.17 Aromatic C-H stretching, 1,703.14 C = O stretching of esters, 1,687.71 C = O stretching of aldehyde, 1,178.51 C-O stretching. 1 HNMR (CDCl3, 400 MHz) δ: 1.717–1.702 (d, 3H, -CH3), 3.930 (s, 6H, 2 × OCH3), 4.179–4.136 (q, 1H, -CH-), 5.299 (s, 1H, Ar-H), 7.431–7.409 (d, 6H, Ar-H), 7.791–7.767 (d, 2H, Ar-H), and 9.914 (s, 1H, -CHO). MS: m/z 365.1250 (M+) (100% abundance).
Solubility and partition coefficient
The result of solubility studies implicated that the synthesized drug was slightly soluble in 0.1 M NaOH. It was insoluble in water and 0.1 M HCl. The synthesized drug displayed moderate-to-high solubility in methanol, ethanol, dichloromethane, chloroform, and benzene. The solubility of the synthesized drug in organic solvents demonstrated lipophilic feature of synthesized drug. It was also presumed that the synthesized drug had good oral absorption due to higher value of Log p. The Log p of the synthesized drug was found to be 5.28 that clearly indicated absorption through the GI tract.
Chemical stability
The synthesized drug showed the resistance in hydrolysis at lower pH while it got hydrolyzed at higher pH gives the idea of its stability. The result obtained implicating that the drug got passed unhydrolyzed through the stomach and showed enough stability to be absorbed intact from the intestine. The calculated rate constant for hydrolysis and the corresponding half-life of the synthesized drug were found to be 2.872 × 10−6 s−1 and 79.78 h at pH 2 and 1.506 × 10−5 s−1 and 41.85 h at pH 7.4, respectively. The reactions were monitored for the decrease in ester concentration versus time and were found to display pseudo-first-order kinetics over several half-lives.
Pharmacological evaluation
The anti-inflammatory, analgesic, and ulcerogenic activities were done for synthesized drug. During anti-inflammatory activity, the Carrageenan-induced paw method utilized Carrageenan (1% w/v) to induce paw edema in control. The paw edema was developed in all groups by injecting Carrageenan (1% w/v in saline) at the subplantar region of the right hind paw. The reduction in inflammation was monitored at regular intervals for 24 h. The perimeter of paw was measured by using Vernier calipers. The result depicted significant inhibition of Carrageenan-induced inflammation (Table 6). The data of anti-inflammatory activity were found to be comparable with the standard drug NAP.
Anti-inflammatory Activity
Data are expressed as mean ± SD of six experiments; —, not determined.
p < 0.0001 extremely significant as compared with control.
p < 0.0001 extremely significant as compared with NAP.
p < 0.001 very significant as compared with NAP.
p < 0.01 significant as compared with NAP.
SD, standard deviation.
The percentage inhibition was calculated as follows:
where Dc = Paw diameter of control
Dt = Paw diameter of treated group
The analgesic activity was measured by the abdominal writhing method. The average number of writhes in each group of mice were calculated. The result obtained at equivalent dose showed considerable retention of analgesic activity (Table 7). The calculated ulcerogenicity data signify that the synthesized drug NVN, had much lesser ulcer index than the parent drug NAP, that is, ∼50% (Table 7). The index was obtained clearly signifying that the free carboxyl group present in parent drug is masked. It may be the cause of ulceration, which is overcome here by this chemical modification.
Analgesic and Ulcerogenic Activity
Data are expressed of six experiments.
p < 0.0001 extremely significant as compared with control.
p < 0.05 significant as compared with NAP.
p < 0.01 significant as compared with NAP.
SEM, standard error of mean; Ui, ulcer index.
Conclusion
Molecular docking simulation-based in silico virtual screening using Autodock was utilized in the current experimental study to design potential COX-2-targeting anti-inflammatory compounds. Three compounds NSES, NQR, and NVN showed promising in silico results with potent inhibition of the COX-2 enzyme, good pharmacokinetic properties, and the absence of any major toxic effects. These molecules can serve as promising lead compounds for further experimental validation as a novel pain-relieving drug.
In the present study, NVN drug was designed, synthesized, and evaluated as safer NSAID. The synthesized derivative was found to be chemically stable and biolabile. The synthesized drug showed desirable anti-inflammatory, analgesic activity with prominent reduced ulcerogenicity at equivalent doses. This may be due to an upgraded physicochemical property requisite for improved bioavailability. This may lead to design a safe and effective drug molecule. On the basis of these observations, it can be concluded that there is advantage of giving NAP and VN in the form of a single molecule, that is, NVN drug.
Footnotes
Acknowledgments
Authors thank Central Instrument Facility, Bose Institute, Annex Building (First floor) Centenary campus, Kolkata for NMR and Mass data. Authors also thank the Management, GLA University, Mathura, UP, India for providing the research facilities and financial assistance to carry out this work at the Institute of Pharmaceutical Research.
Disclosure Statement
No competing financial interests exist.
Funding Information
The project was funded by GLA University, Mathura, UP, India. (No. GLAU/RO/R&D/5560/16).