
Abstract
In this study, we prepared gelatin-coated mesoporous hollow silica nanospheres (GSN) as a drug carrier to improve the water solubility and regulate the release rate of glimepiride (GLM). GLM was loaded into GSN by an absorption method, and drug-loaded samples (GLM-GSN) were characterized by differential scanning calorimeter (DSC) and X-ray diffraction (XRD). Cellular uptake and in vivo intestinal uptake experiments were performed in rats. In addition, the studies of in-vitro drug dissolution, pharmacokinetics, and pharmacodynamic experiments also were performed. GLM-GSN showed excellent drug loading (39.7% ± 0.7%) and sustained GLM release. The state of GLM in GSN was amorphous according to DSC and XRD results. Cellular uptake and in vivo intestinal uptake experiments indicated that GSN could be effectively absorbed, and an MTT experiment demonstrated that GSN had good biocompatibility. Furthermore, the GLM-GSN had a higher bioavailability in pharmacokinetics experiments and a prominent hypoglycemic effect on type-2 diabetes model rats in pharmacodynamic experiments. This study clearly shows that GSN is a promising platform for delivering GLM for the treatment of type-2 diabetes.
Introduction
Diabetes is a disease that involves metabolic abnormalities. Due to impairment of islet cell function, hyperglycemia occurs in all diabetic patients worldwide. The type of diabetes with the highest prevalence is noninsulin-dependent diabetes. Glimepiride (GLM), as a third-generation sulphonylurea drug, has been widely applied for type 2 diabetes treatment. 1 –3 However, GLM is a typical BCS II drug that has poor water solubility. Thus, improving its solubility and dissolution rate in the gastrointestinal tract is crucial for enhancing its oral bioavailability and clinical efficacy. Moreover, the half-life of GLM is ∼5 h, which is too short for long-term therapy. Hence, it is necessary to design a drug delivery system that can increase the solubility of GLM and adjust its release rate to obtain a sustained release effect. Currently, numerous approaches are being used to solve this problem. Ahmed et al. developed a transdermal GLM delivery system using ethosome nanovesicles that could control drug release. 4 Ahmed et al. prepared a self-emulsifying GLM nanoemulsion for .transdermal patches and improved drug bioavailability. 5 Bera et al. designed carboxymethyl fenugreek galactomannan-gellan gum-calcium silicate composite beads for controlled GLM delivery, and the system showed excellent mucoadhesive properties and good hypoglycemic effects. 6 With the development of nanotechnology, mesoporous materials, such as mesoporous silica 7 –11 and mesoporous carbon, 12 –15 provide a new method to improve the water solubility of insoluble drugs. These materials have a large specific surface area, large pore volume, and nanoscale aperture. Taking advantage of these structural advantages can allow high dispersion of insoluble drugs, reduce the size and crystallinity of drug particles, and improve water solubility. Furthermore, the surface of mesoporous materials is easy to modify with amino or carboxyl groups, which can bond with the macromolecule polymer, and surface grafting can be used to regulate the rate of drug release.
Gelatin, as a natural biopolymer, is obtained from the partial hydrolysis of collagens. Its safety as a pharmaceutical excipient is generally recognized by official institutions in various countries. In addition, it is also considered as a preeminent drug delivery material in formulations such as microspheres, 16 microcapsules, 17 and three-dimensional ordered nanomaterials. 18 In this study, mesoporous hollow silica nanospheres were prepared by a surfactant-free synthesis method and, due to the space restriction effect of the mesoporous hollow structure, the dissolution rate of GLM was significantly improved. When gelatin-coated mesoporous hollow silica nanospheres (GSN) were used, the release rate of GLM was effectively regulated, and a sustained release effect was obtained. Then, in vitro characterization (differential scanning calorimeter [DSC], X-ray diffraction [XRD]), in vivo pharmacokinetics, and pharmacodynamics tests were conducted to determine whether GSN is a suitable potential carrier material for GLM.
Materials and Methods
Materials and Animals
Gelatin was purchased from Aladdin Reagent Company with gel strength 250 g bloom, and [2-(acryloyloxy)ethyl] trimethylammonium chloride (DAC, 80 wt%) was provided by Jinan Wanduoxin Chemical Co., Ltd. (Jinan, China). Styrene, ethanol, tetrahydrofuran, ammonia, cetyltrimethylammonium bromide (CTAB), tetraethyl orthosilicate (TEOS), 2,2′-azobis(2-methylpropionamide)dihydrochloride (AIBA), gelatin, chloroform, and sodium dodecyl sulfate (SDS) were supplied by Tianjin Yongyi Fine Chemical Co., Ltd. (Tianjin, China). RPMI 1640 medium, fetal bovine serum, Caco-2 cells, propidium iodide, annexin V-FITC, dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT), paraformaldehyde, streptozotocin, and trypsin were obtained from Beijing Dingguo Changsheng Biotech Co., Ltd. (Beijing, China).
Synthesis of GSN
Mesoporous hollow silica nanospheres (MSN) were synthesized based on a study reported by Qiu et al. 19 In brief, styrene (40 g) was added to a water solution (390 mL) containing 1 g of DAC under vigorous stirring for 30 min. Then, 0.1 g/mL of AIBA water solution (10 mL) was added to the above system under nitrogen and stirred at 90°C for 24 h. A polystyrene nanosphere emulsion was obtained. Distilled water, ethanol, ammonia, and CTAB in a mass ratio of 29:12:1:0.8 were mixed, and the above polystyrene nanosphere emulsion (10 g) was dispersed in the mixed solution under stirring. Then, 4 g of TEOS was added dropwise to the suspension. The precipitate that was obtained by centrifugation was dispersed into tetrahydrofuran to remove the template. The obtained mesoporous hollow silica nanospheres were washed with ethanol and dried in a vacuum. Then, 100 mg of mesoporous hollow silica nanospheres were dispersed in 10 mL of gelatin solution (1 mg/mL) under stirring. After 1 h, the obtained GSN were centrifuged, dehydrated with ethanol, and dried in a vacuum.
Drug Loading
GLM was loaded into GSN by an adsorption method. 20 A total of 25 mg of GLM was dissolved in 1 mL of chloroform, and 50 mg of GSN was dispersed in the above GLM solution under stirring for 5 h. The sample (GLM-GSN) was centrifuged and dried under vacuum. The drug content (DC) of GLM-GSN was determined by high-performance liquid chromatography (HPLC, LC-2030; Shimadzu, Inc., Japan) at 228 nm and calculated by the following equation: DC (%) = Amount of drug in GLM-GSN/Amount of GLM-GSN × 100%. The mobile phase was a mixed solvent composed of acetonitrile and sodium dihydrogen phosphate solution (pH 4, 0.1%) at a volume ratio of 50:50. As a control, mesoporous silica was subjected to the same drug-loading process as described above, and the sample was called GLM-MSN.
Characterization of GSN and GLM-GSN
The morphological structures of mesoporous hollow silica nanospheres were observed by scanning electron microscopy (SEM; JEOL JSM-7001F, Japan) transmission electron microscopy (TEM, Tecnai G2F30; FEI) and the particle size was determined with laser particle sizer (Nano-ZS90; Malvern, United Kingdom). The gelatin content in GSN was measured by thermal gravimetric analysis (TGA). The state of GLM was detected by a DSC-60 (Shimadzu, Inc.) and XRD (Rigaku Denki, Japan).
Drug Release
A drug release test was performed according to the paddle method by an intelligent dissolution instrument (RC806D; Tianda Tianfa, China). The release medium was 900 mL of phosphate-buffered saline (PBS), pH 7.8. The temperature was 37°C ± 0.5°C, and the speed of the agitator was 100 rpm. The GLM-GSN (equivalent to 2 mg of GLM) was dispersed in the release medium, and at a predetermined time, the DC of the samples was measured by the HPLC method.
GSN Cytotoxicity, Cell Uptake, and Intestinal Mucosal Uptake
An MTT assay was used to evaluate the biosafety of GSN as an oral drug carrier material. Caco-2 cells were seeded onto 96-well plates. The cytotoxicity of GSN was measured in Caco2 cell monolayers by exposure to a GSN suspension at concentrations of 0–500 μg/mL for 48 h. MTT solution was added to each well, and the plates were incubated for 4 h. Next, DMSO was added, and the plates were incubated for 10 min. A cytotoxicity assay of GSN was performed using a plate reader (DNM-9606) at 492 nm. 21
GSN (100 mg) were dispersed at 1 mg/mL of fluorescein isothiocyanate (FITC) anhydrous ethanol solution under stirring for 4 h, and then FITC-labeled GSN (FITC-GSN) were obtained by centrifugation. The sample was stored in a vacuum. Caco-2 cells were seeded in six-well plates at a density of 5 × 104. After the cells had attached, FITC-GSN (50 μg/mL) was added to six-well plates and incubated for 1, 2, and 3 h. After fixation in 4% formaldehyde, the cells were incubated in a 0.1% Triton X-100 PBS solution containing 10% bovine serum albumin for 30 min. The Caco-2 cells were stained with Hoechst 33342 PBS solution and rhodamine-phalloidin PBS solution in order. Finally, the cell uptake of FITC-GSN was observed by laser scanning confocal microscopy (LSCM; Leica, Germany).
The intestinal distribution of FITC-GSN was observed by LSCM. Male Sprague-Dawley (SD) rats (∼220 g) were orally administered 10 mg/kg FITC-GSN and sacrificed after 1 h. The duodenum, jejunum, and ileum (∼1 cm) were excised and fixed with 4% paraformaldehyde after washing with PBS. After gradient dehydration in 15%, 20%, and 30% sucrose, the samples were fixed with cryoembedding media (OCT) under freezing conditions. After frozen sectioning, the slices were stained with Hoechst 33342 and rhodamine-phalloidin. Each segment was observed by LSCM.
Pharmacodynamic Evaluation
Forty-five SD rats were fed a normal diet for 1 week. Eight were randomly selected as the normal control group, and the remaining rats were used as diabetic model animals. The normal control group was fed a normal diet, and the diabetic model group was fed a high-fat and high-sugar diet for 8 weeks. Then, the type 2 diabetes model was induced by a single intraperitoneal injection of streptozotocin (25 mg/kg). After 72 h, the blood glucose level (BGL) was measured via the tail, and measurements were obtained continuously for 3 days. Rats with BGLs that exceeded 11.1 mmol/L were selected as diabetic models.
The model group was randomly divided into three groups. The first group was given citrate buffer each day, the second group was given a marketing preparation (GLM capsules from Sichuan Purdue pharmaceutical factory) each day (2 mg GLM/kg), and the third group was given GLM-GSN (2 mg GLM/kg) by oral gavage each day for 8 weeks. Normal rats were used as controls. Fasting blood glucose was measured using a commercial glucose kit by the tail-cutting method at specific time points. The body weight and food intake of each group were monitored daily. After 8 weeks, serum lipid level, including total cholesterol, triglyceride, and free fatty acid (FFA), was determined by the oxidase method. The skeletal muscle tissues were stripped, and the western blot method was used to detect the expression level of glucose transporter 4 (GLUT4) protein. The protein was extracted from the skeletal muscle tissue by ultrasonic comminution, and the protein concentration was determined by the bicinchoninic acid (BCA) method. The protein content of each group was adjusted to a specific concentration, and the protein was denatured by boiling. Samples (20 μL) and standard protein were obtained from each group for polyacrylamide gel electrophoresis. After the gel was transferred, 5% skimmed milk powder was used to block the polyvinylidene fluoride membrane for 1 h. After cutting, the membranes were incubated at 4°C with the primary antibody overnight and then incubated with secondary antibody for 2 h. After enhanced chemiluminescence reagent staining, the imprinted zone was quantitatively analyzed by a UVP system (UVP; LLC., Upland, CA). The liver tissues from each group were fixed with 4% paraformaldehyde solution for 48 h and then dehydrated with different concentrations of ethanol. After paraffin embedding and sectioning, the obtained tissue slice was stained with hematoxylin-eosin (HE), and the histopathological changes were observed under an optical microscope.
Pharmacokinetics Evaluation
According to the Animal Protection and Utilization Committee of Jinzhou medical university, six rabbits (2.5 ± 0.5 kg) provided by the experimental animal center of Jinzhou Medical University were randomly divided into two groups and used to pharmacokinetics study. After fasted for 12 h, the marketing preparation and GLM-GSN (2 mg GLM/kg) were administrated by oral gavage. Blood samples (3 mL) were collected at the specified time point from the ear vein and centrifuged at 4,000 r/min for 5 min, then stored at −20°C. Two milliliters methanol was added to 1 mL serum sample. After vortexed for 30 s, the mixture was centrifuged at 10,000 r/min for 15 min. One hundred fifty microliters hydrochloric acid solution (0.3 mol/L) was added to the obtained supernatant samples and the mixture was vortexed for 30 s. Then 4 mL of a mixed solvent composed of dichloromethane and n-hexane (3:1, v/v) was added above system. The organic layer was separated by centrifugation after vortexed for 1 min and blown dry under nitrogen flow. One hundred microliters methanol was used to redissolve the dried samples and 50 μL was injected into HPLC (Shimadzu LC-2030) with a UV–vis detector at 228 nm using a C18 Agilent column (150 mm × 4.6 mm, 5 μm). The mobile phase was the mixture of acetonitrile and 0.1% sodium dihydrogen phosphate solution (50:50, V/V) and the flow rate was 1 mL/min. The maximum plasma concentration (Cmax), the half-time (T1/2), the time to reach the maximum plasma concentration (Tmax), and the area under the plasma concentration-time curve (AUC) were calculated by PKSolver software. 22
Statistical Analysis
All experimental data are expressed as the mean ± standard deviation. The difference between the groups was analyzed by analysis of variance and Bonferroni tests using SPSS 17.0 software and was statistically significant (P < 0.05).
Results
Morphology and Structure
The MSN were prepared using a template method. The SEM and TEM images in Figure 1A and B indicate that the MSN had good dispersibility with a particle size of 140 nm. The results (140 ± 5 nm) from laser particle sizer were consistent with those from electron microscopes. The unique mesoporous shell structure and the hollow structure with a diameter of 120 nm made the MSN an outstanding candidate for a drug delivery carrier. To avoid drug recrystallization caused by rapid dissolution, a gelatin coating was used to regulate drug release. After coated with gelatin, the particle size was 154 ± 7 nm. The gelatin content used to coat the surface of the MSN was determined by TGA. The result is shown in Figure 1C, the gelatin coating comprised ∼7% of GSN weight. The adsorption method was applied for GLM loading and the drug loading was determined by HPLC. Finally, the DC of GLM-GSN was determined to be 39.7% ± 0.7%. The high drug-loading capacity and low density indicated GSNs as a suitable insoluble drug carrier material.
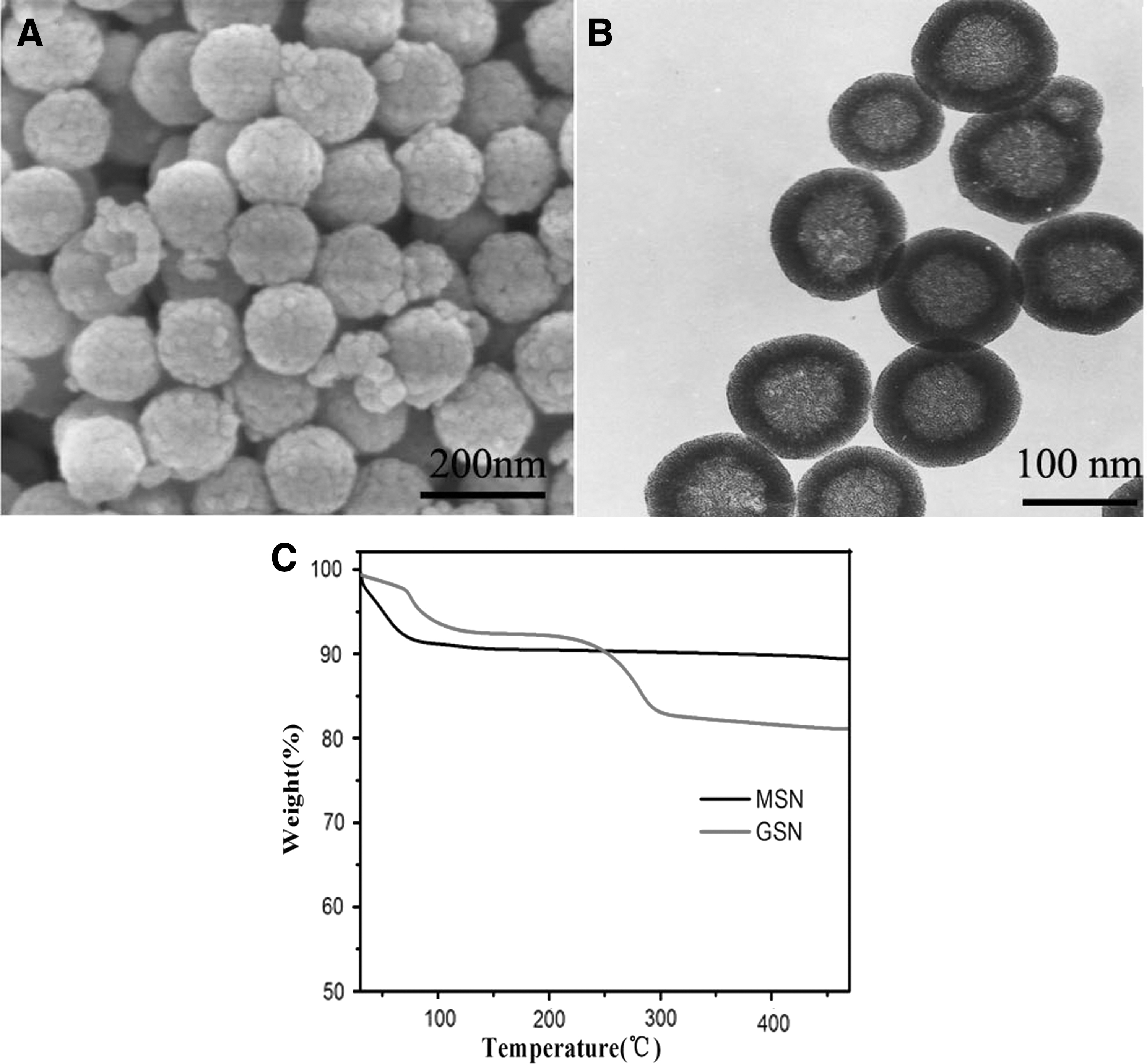
SEM
Solid State Characterization
The results from XRD and DSC indicated that GLM was in an amorphous state in GSN. 6 As shown by Figure 2A, the XRD profile of GSN showed no characteristic diffraction peak. GLM exhibited a series of characteristic crystal diffraction patterns, indicating that it was in a crystalline state. Compared to the physical mixture (PM) with the same proportion, GLM-GSN had no diffraction peak for GLM. This demonstrated that the loaded GLM was not in a crystal form. The above findings were further verified through DSC. In Figure 2B, both GLM and PM exhibit an endothermic peak at 215°C, which was attributed to the melting and endothermic reaction of GLM. The DSC profile of GSN was a smooth straight line, indicating no phase transformation. GLM-GSN did not show any endothermic or exothermic processes. These results demonstrated that the GLM in GLM-GSNs was amorphous. 23
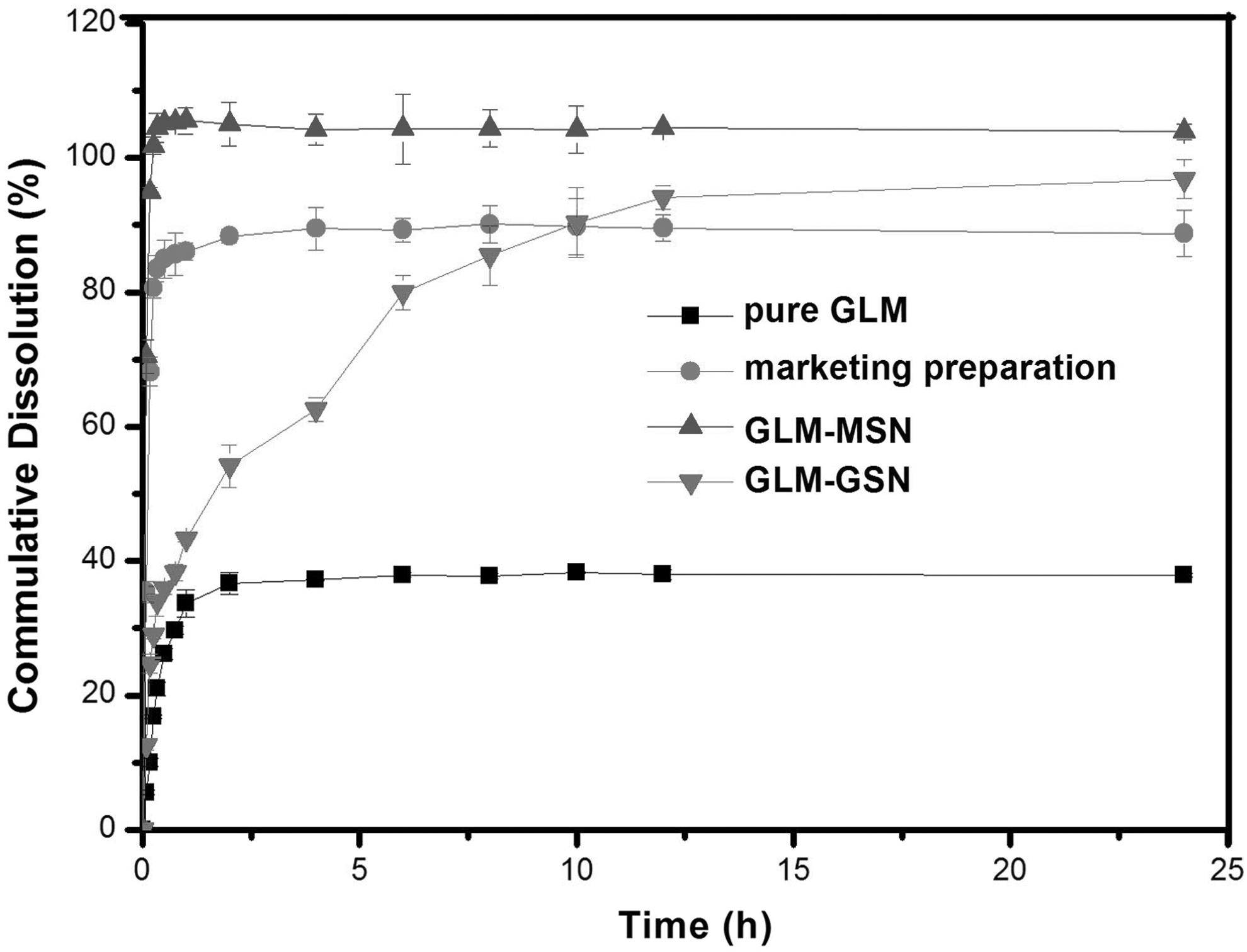
In vitro release curves of GLM, marketing preparation, GLM-MSN and GLM-GSN (n = 3). GLM, glimepiride.
In Vitro Drug Dissolution
The measurement results of the dissolution test confirmed that the mesoporous characteristics of mesoporous hollow silica nanospheres can significantly improve the dissolution of GLM. As shown in Figure 3, pure GLM presents low dissolution (33.6% ± 2.01%) at 1 h. The marketing preparation showed 86.5% ± 1.28% of drug dissolution at 1 h. By comparison, the GLM-MSN showed 80.6% ± 1.51% drug dissolution at 15 min. GLM-GSN showed about 40% of drug release at 1 h and more than 90% drug release at 24 h.
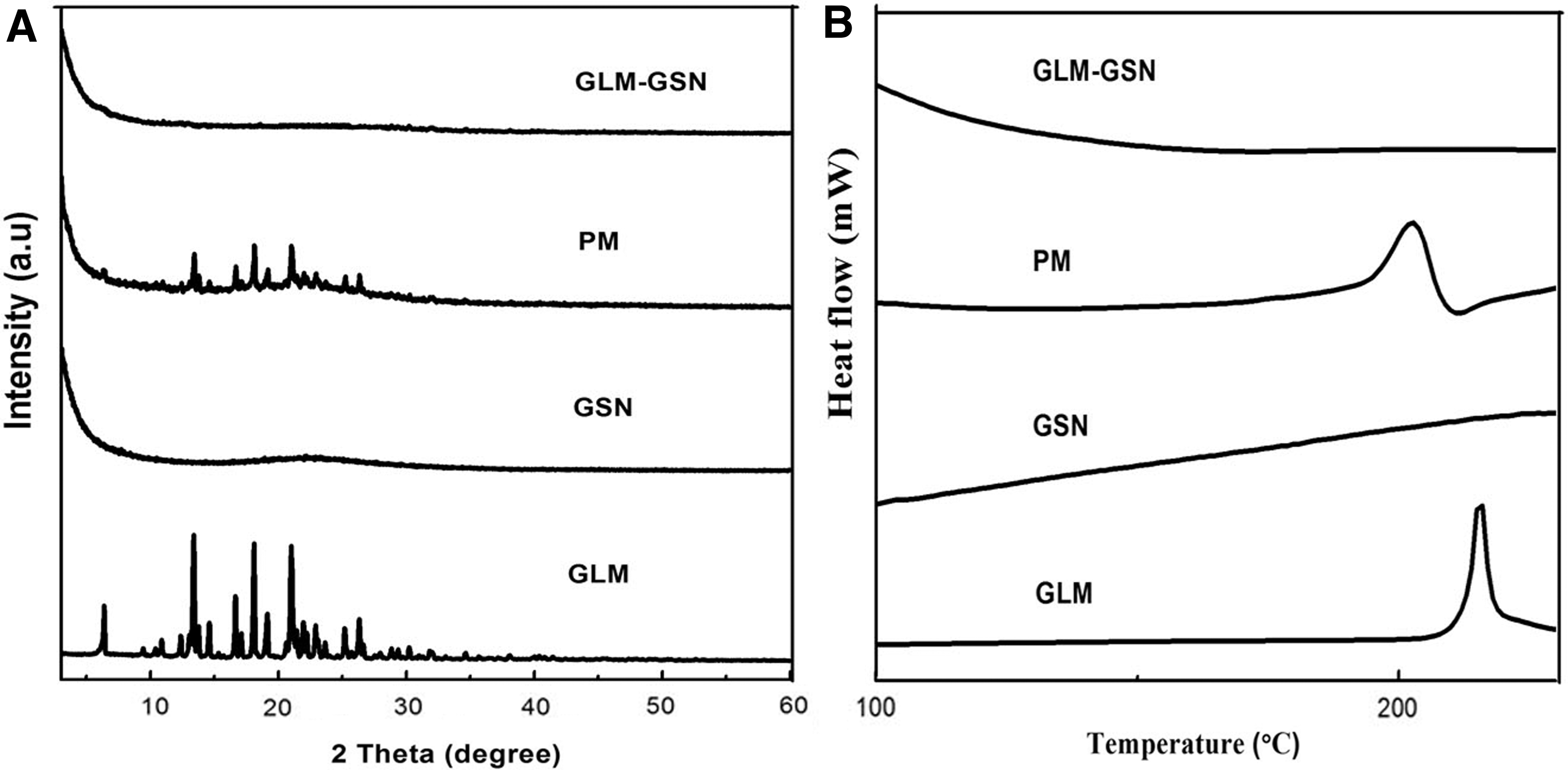
The X-ray diffraction
In Vitro Cytotoxicity Assay of Carriers and Drug Uptake Study
An MTT assay was used to investigate the cytotoxicity of GSN in the colon cancer cell line Caco-2. As shown in Figure 4A, the cell viability was greater than 90% for each concentration of GSN. This indicated that GSN had good biological safety. The cell uptake study results demonstrated that FITC-GSN can be efficiently taken up by Caco-2 cells. Figure 4B and C show LSCM images of Caco-2 cells treated with FITC-GSN at 2 h. In Caco-2 cells, green fluorescence spots appeared after the cells were treated for 2 h, indicating that FITC-GSN could be internalized into the cytoplasm via the cell membrane and the uptake of carriers by cells was time dependent. LSCM images of GSN transported across the intestinal epithelium are shown in Figure 4D. The section images of the jejunum and ileum at 2 h after oral administration of FITC-GSN showed strong green fluorescence compared to those of the duodenum, indicating significantly increased transepithelial transport in the villi and epithelial cells. 24 These results showed that GSN can significantly improve drug absorption through nanoscale effects.
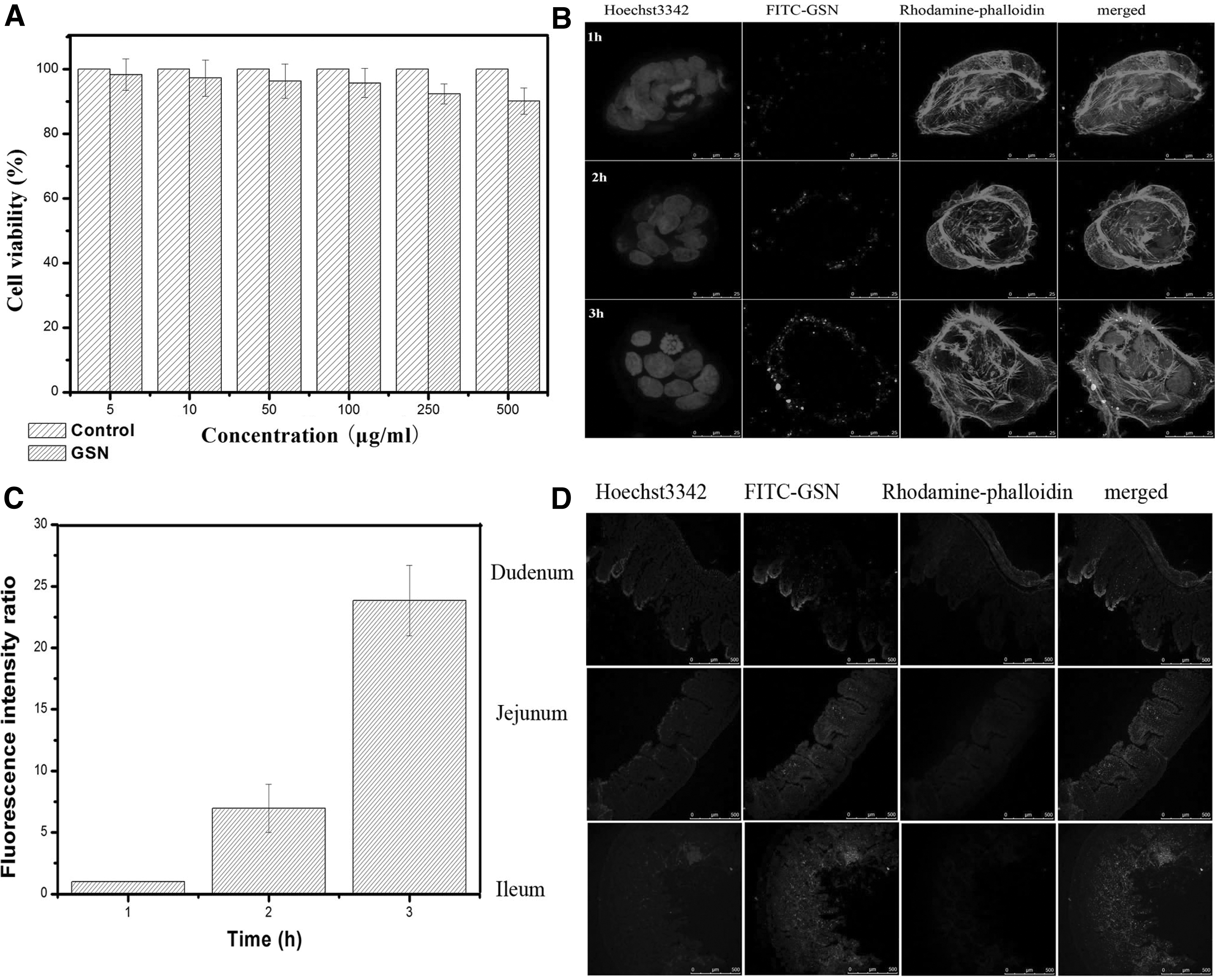
Cell viability
GLM-GSN Ameliorate Symptoms in Diet-Induced Diabetic Mice
Due to injury and destruction of streptozotocin, high-fat, and high-sugar diet to pancreas, BGL significantly elevated. The antihyperglycemic activity of the GLM-GSN and a marketing product were evaluated in diabetic animals using acute and chronic dosing regimens. As shown in Figure 5A, a single dose of the marketing product produced a sharp decrease in the BGL that reached a maximum value at 2 h. In comparison, the GLM-GSN group exhibited a moderate decrease in BGL and a better BGL reduction effect that lasted for more than 24 h. In Figure 5B, chronic administration of GLM-GSN (BGL 9.58 ± 0.29) shows a steady hypoglycemic effect, which was better than that observed in the group that received the marketing product group (BGL 11.41 ± 0.38). The weight and food intake results further supported the above findings. In Table 1, compared to the normal group, the weight of the diabetic group decreased significantly and the intake of food increased significantly. The GLM-GSN group and the marketing product group exhibited obvious improvement in body weight and reduced food intake. The GLM-GSN group showed a better effect. The western blot experiments further confirmed the above results. The western blot analysis results in Figure 5C and D show that GLUT4 was significantly lower in skeletal muscle cells of type 2 diabetic rats than in the corresponding cells of normal rats, and the expression of GLUT4 was significantly increased after administration of GLM-GSN and the marketing product. The GLM-GSN group exhibited greater GLUT4 expression.

Hypoglycemic effect of after single-dose administration
Changes in Body Weight, Food Intake, and Serum Lipid
The results are expressed as mean ± standard deviation. There were significant differences among all groups, p < 0.05.
GLM, glimepiride; GSN, gelatin-coated mesoporous hollow silica nanospheres.
In Table 1, the serum lipid level of the model group increased significantly compared with the normal group. GLM-GSN group and the marketing product group improved the serum lipid level and GLM-GSN group had better effect. HE staining of liver tissue further verified the above results. In Figure 6, it was found by optical microscope that the structure of the liver lobule in the normal control group was regular, the hepatocyte cords was arranged radially and orderly around the central vein, the size of hepatocyte was uniform, and the cytoplasm was rich. By comparison, the structure of the liver lobule in the diabetic model group was unclear, and the arrangement of hepatocyte cord was disorder. The hepatocyte appeared steatosis, multifocal, and small necrosis. The inflammatory cell infiltration happened in the portal area. Although the liver still showed a certain degree of damage compared to the normal group, the liver cell cords and sinusoids of the GLM-GSN group and the marketing preparation group were relatively arranged regularly and the central vein structure was complete nearly. These results illustrated that GSN were excellent carriers for GLM.
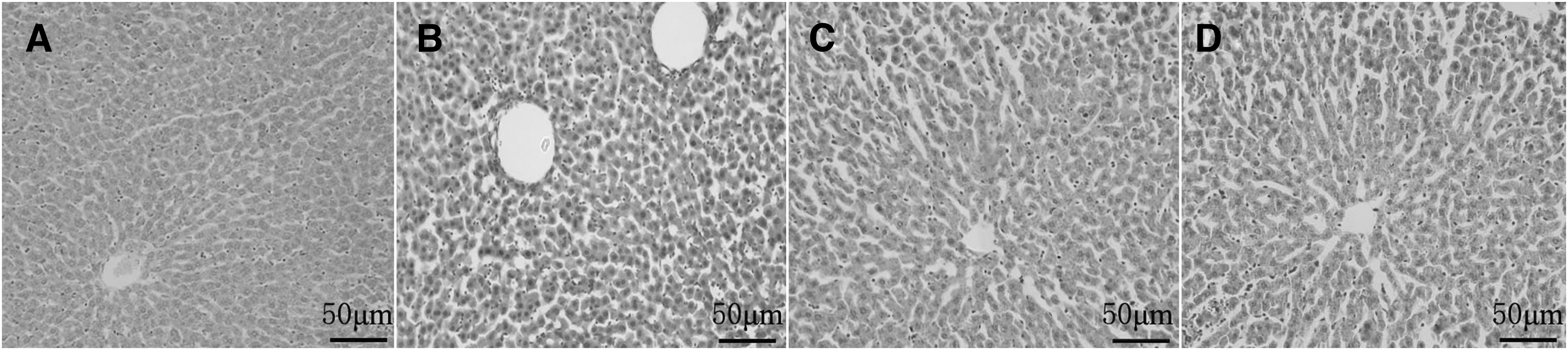
Hematoxylin-eosin staining pictures of liver tissue sections. (
Pharmacokinetics Study
The plasma concentration-time curves of the marketing preparation group and GLM-GSN group are shown in Figure 7. The pharmacokinetic parameters are exhibited in Table 2. Compared with the marketing preparation, Tmax and MRT of GLM-GSN had significantly longer time and the AUC increased obviously. The relative bioavailability of GLM-GSN was 162.9%. The sustained drug release of GLM-GSN enhanced GLM absorption, which promoted the improvement of bioavailability and also was helpful to minimize side effects and improve the therapeutic effect.
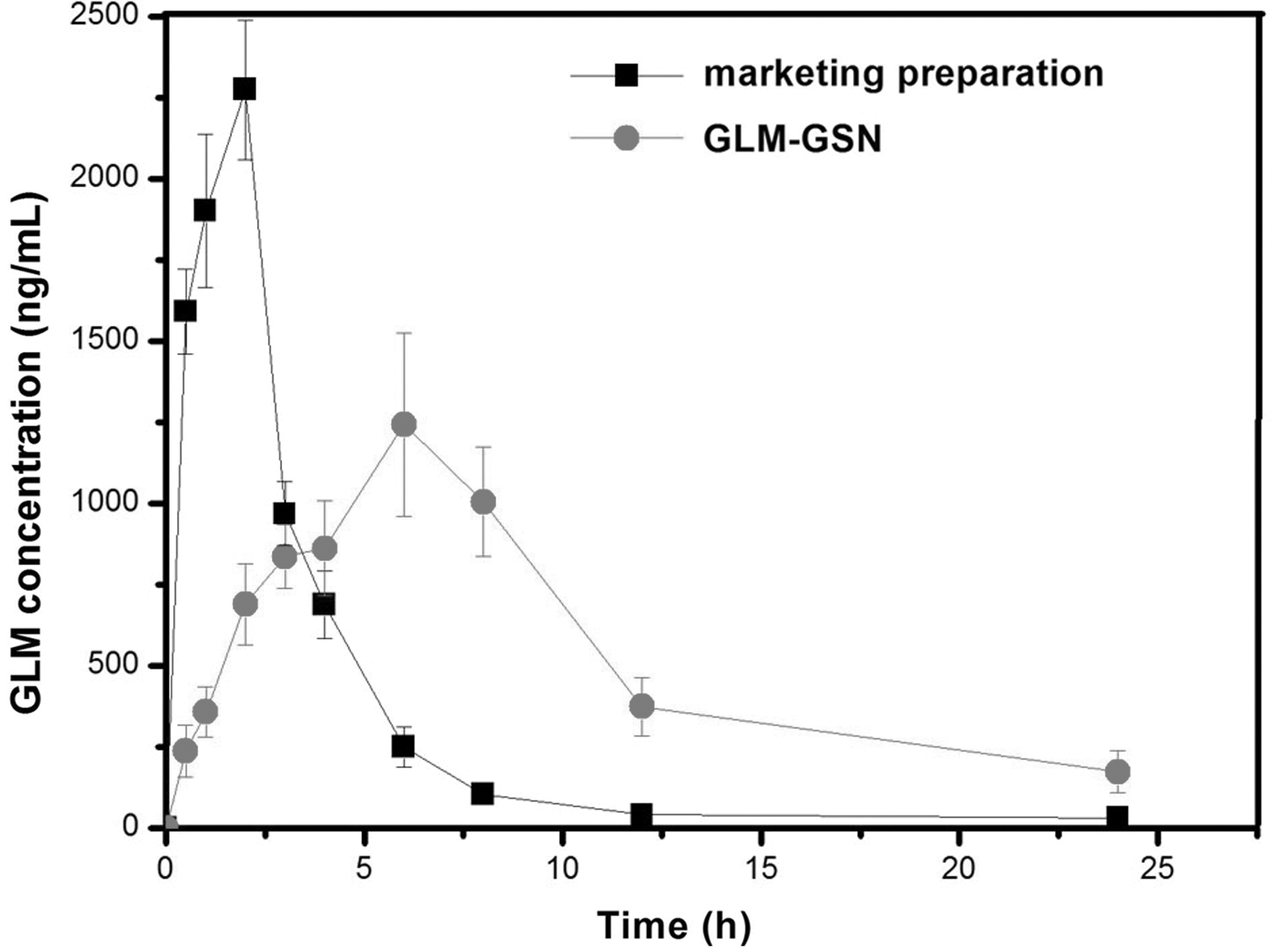
The plasma drug concentration-time curves of the marketing preparation and GLM-GSN.
Pharmacokinetic Parameters
p < 0.05.
AUC, area under the plasma concentration–time curve; Cmax, maximum plasma concentration; MRT, mean residence time; Tmax, time to reach Cmax.
Discussion
Structure, Drug State, and Release
The nanoscale effect of the mesoporous structure was thought to be responsible for improving the dissolution rate. The nanoscale pore structure of MSN can limit the crystallinity of insoluble drugs, decrease in drug particle size, and produce an amorphous state, which is directly related to the improvement of dissolution. The highly dispersed nanodrugs increased its contact with the dissolution medium and increased the speed of dissolution. This was propitious for increasing the absorption and improving the efficacy of the drug. However, the accelerated dissolution of insoluble drugs might cause recrystallization of dissolved drugs, which is not conducive to drug absorption. To avoid this, encapsulated gelatin was used, which played a role in regulating drug release. Due to the blocking effect of gelatin network,
Drug Absorption and Bioavailability
According to the results of cell uptake and intestinal mucosal uptake by duodenum, jejunum, and ileum sections, GSN showed excellent transmembrane capability. GLM-GSN into cell or mucous membrane produced the accumulation and released the drug continuously, which should promote the drug to be absorbed in large quantities into the systemic circulation and improve the drug bioavailability. The results of pharmacokinetic experiments demonstrated that GSN could surely regulate the release rate of GLM, resulting in a sustained plasma concentration, and GLM-GSN could improve the oral bioavailability of GLM compared to the marketing preparation. For the marketing preparation, the amounts of GLM released rapidly in a short time could not only achieve a large Cmax subsequently but also cause the drug precipitation in the GI tract, which influenced the GLM absorption. However, the sustained release characteristics of GLM-GSN from 0 to 24 h obtained a satisfied oral bioavailability with a long-playing plasma therapeutic concentration. The feasibility and validity of the gelatin-coating strategy in this study confirmed that GSN had good solubilization effect and could release drugs slowly, which was the dominant factor enhancing the GLM oral bioavailability.
Hypoglycemic Effect
The fast hypoglycemic effect of the marketing product resulted in a short duration of action, and blood glucose rebound could easily occur during the interval of drug administration. The BGLs fluctuated considerably, which can cause side effects and potentially lead to liver damage. 25 The hypoglycemic effect of the GLM-GSN group was moderate, and a long-lasting effect was observed, which did not cause blood sugar glucose fluctuations, resulting in a safer hypoglycemic action. In terms of BGL, weight, and food intake, long-term administration results confirmed the above conclusion. GLM-GSN significantly improved the symptoms of diabetes. GLUT4 is the main protein that assists adipocytes and skeletal muscle cells in glucose transport. In a normal state, GLUT4 is distributed in various organelles, including the mitochondria, Golgi, and sarcoplasmic reticulum. It translocates to the cell membrane under insulin or exercise stimulation, and glucose molecules are loaded from high-concentration to low-concentration regions. The GLUT4 content of the cells of type 2 diabetic rats is relatively low, and their glucose uptake ability is also poor mainly due to impaired GLUT4 membrane transport. The expression results of GLUT4 further validated the role of GLM-GSN.
Fatty liver is closely related to type 2 diabetes. Type 2 diabetes is often associated with insulin resistance and liver as the target organs of insulin resistance reduce the glucose intake of the liver, which leads to decrease of the liver's ability to oxidize FFAs. Long-term fat accumulation in the liver can cause fatty liver and liver damage. 26,27 In controlling the serum lipid level, GLM-GSN group showed better results. The HE staining results of liver tissue sections also corroborated the above results. Therefore, based on the results of pharmacokinetics and pharmacodynamics, we have concluded that GLM-GSN has a good hypoglycemic effect.
Conclusion
This investigation showed that GSN have good potential as drug delivery carriers of insoluble hypoglycemic drugs, have a high drug-loading capacity, and allow sustained drug release. GLM-GSN were successfully prepared and characterized by TGA, DSC, and XRD. An intake experiment indicated that GSN could increase drug absorption due to nanoscale effects and good enhanced membrane adhesion produced by gelatin. Pharmacodynamic studies demonstrated that GLM-GSN showed significant hypoglycemic potential compared to the marketing product in an alloxan-induced diabetic rat model. In summary, the sustained and prolonged effect of GLM-GSN can reduce drug dosage and frequency of administration and result in better patient compliance.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by National Natural Science Foundation of China (No. 81770459 and No. 81970369) and Natural Science Foundation Guidance Plan of Liaoning Province (No. 201602301).