
Abstract
Diclofenac sodium (DS) is categorized under the nonsteroidal anti-inflammatory class of drugs that also belongs to biopharmaceutical classification system (BCS) class II. It has limited dissolution parameters which also resist the total bioavailability but it has a good transdermal permeability characteristic and the pharmacokinetic parameters of DS make it suitable for the formulation of nanostructured-lipid carrier (NLC)-based gel transdermal delivery. The research aimed to design and develop a drug-delivery system (DDS), i.e., DS-NLCs incorporated in gel to modulate its anti-inflammatory action via skin. The formulation was optimized using Taguchi’s approach and the resultant NLCs were thoroughly characterized, including assessments for zeta potential, particle size, and morphological evaluation. Furthermore, particular investigations were carried out for DS-NLCs, including drug encapsulation efficiency, ex vivo release properties in Phosphate Buffer Saline at pH 7.4, and an in vivo skin irritation test. DS-NLCs had a mean size of 339 ± 25 nm and were spherical-shaped particles. With an encapsulation effectiveness of 84%, the NLCs were found to have effectively loaded drugs. Moreover, these NLCs demonstrated a sustained release characteristic that persisted for a maximum of 24 h, suggesting their potential for gradual and regulated drug release. Lipid components demonstrated good stability over 90 days and were biocompatible with the DS. Furthermore, compared with the usual formulation, topical gel loaded with NLC (GNLC) containing DS considerably suppresses edema in the in vivo result, suggesting that the developed formulation has superior anti-inflammatory efficacy. These NLCs provide prolonged release and better drug solubility, both of which boost therapeutic outcomes and control the drug’s anti-inflammatory potential. The study’s conclusion emphasizes DS-NLC’s potential as a cutting edge and effective medication delivery technology. The results indicate the need for more preclinical research, which presents an effective direction for developing a more potent and well-tolerated therapeutic strategy.

INTRODUCTION
Diclofenac sodium (DS) is a nonsteroidal anti-inflammatory drug that treats inflammatory disorders and helpful for pain management, including arthritis, with good patient compliance. Due to substantial hepatic metabolism, DS has a limited biological half-life of 1–2 h, requiring frequent high doses.1,2 On the other hand, gastrointestinal tract adverse effects such as internal bleeding, ulcers, or intestinal wall perforation have been linked to DS. 3 Therefore, it is hypothesized that controlled transdermal drug delivery is advantageous over oral or injectable administrations by avoiding first pass metabolism and pain minimization.4,5 The skin rigidity still limits transdermal drug administration in practice. Microorganisms and other external stresses including heat, chemicals, toxicants, and dehydration are among the hazards that the skin layers shield the body against. 6
The research goal has been to produce topical transdermal drug-delivery systems (DDS) by overcoming this barrier based on a thorough understanding of the skin. In this regard, lipid-nanoparticles act as the most effective approaches.7,8 Lipid nanocarriers’ reduced size may increase the specific surface area for absorption of drug transdermally, leading to increased effect of drug. 9 Additionally, transepidermal water loss is decreased by the occlusive action of film development on the skin’s surface, which improves drug permeation via the stratum corneum. 10 Particle size (PS) has been discovered to have an impact on transdermal distribution; liposomes with sizes of 120–810 nm were found to fix in the skin more readily than bigger particles. 11 Additionally, it has been observed that utilizing nanoemulsions (35–68 nm), smaller particles improve drug delivery. 12 A different study reported on the performance of 50–500 nm particles made of the same carboxylate-based nanoparticles. Their intriguing results show that 500 nm particles give more effective drug delivery at later times, while <50 nm particles initially show a high level of the drug. 13 When combined, these results show that the effects of size on skin-drug transport are intricate and contingent upon physiological and anatomical factors.
Nanostructured lipid carriers (NLC) are a type of nano-sized lipid-based particulate vesicle, consisting of biocompatible lipids with a size range of 10–1,000 nm. 14 Numerous chemotherapeutic compounds, including as paclitaxel, 15 doxorubicin or DOX, 16 curcumin, 17 and tamoxifen, 18 are delivered by NLC. A few anticancer drugs, such as doxorubicin, oxaliplatin, vincristine, and irinotecan, have been chosen for clinical trials or are commercially accessible for use in clinical settings. DOX’s lipid-based nanoformulation was among the first drugs to be utilized against cancer on a commercial scale. 19 Because physiological lipids are used to make NLCs, they exhibit good control over drug release and reduce the likelihood that the drug will rupture in biological systems, making them easily tolerated. 20 As was previously mentioned, these are improved versions of solid lipid nanoparticles (SLN) with sophisticated functionality.21,22 Cannabidiol NLCs were synthesized to improve chemical stability and anti-inflammatory activity when it was used in dermal delivery, 23 nimesulide lipid carrier, 24 mometasone furoate SLNs for topical delivery, 25 SLNs and NLCs of sildenafil citrate were prepared to improve transdermal permeation. 26 Dutasteride- 27 and Docetaxel- 28 loaded NLCs were also formulated for improved drug release when these were used on topical routes. The acitretin-loaded gel was prepared for the effective treatment of psoriasis through a topical route. 29 Diclofenac sodium 2-(2,6-Dichloroanilino) phenyl] acetic acid is approved as an anti-inflammatory drug. DS inhibits prostaglandin synthesis by inhibiting the transiently expressed prostaglandin-endoperoxide synthetase also known as cycloxygenase-2. In this research work, an NLC of DS was designed, fabricated, and loaded into a gel-based DDS for delivery across the skin or transdermal route. Taguchi design was implemented for optimization of NLCs, as it is reported as an effective method for optimization by various process parameters including drug and lipid ratio, concentration of surfactant, stirring time, and sonication time. It plays a significant role in suggesting the optimized formulation that significantly influences the PS, polydispersity index (PDI), zeta potential, drug loading (%DL) and encapsulation efficiency (%EE). This process optimization helps in the development of scalable, repeatable or reproducible, and effective DDS which maintains consistent therapeutic results.30–32
The almonds, Oleum amygdalae, are the source of almond oil (AO). Almonds and the oil they yield offer numerous benefits, such as anti-inflammatory, immune stimulating, and antihepatotoxic effects, despite the lack of clear scientific evidence. 33 The Food and Drug Administration (FDA) has approved AO for use on humans and it is deemed safe when applied topically. 34 As a lipid-phase component, AO is used in several drug carriers, including lipid-based systems like microemulsions and nanoemulsions.35–37 Notably, no report in the literature currently in existence combines lipid-based systems—more precisely, NLCs (AO with DS). Pluronic F-68 is a triblock copolymer composed of polyethylene oxide: polypropylene oxide: polyethylene oxide (70:30:70) and it helps to possess more hydrophilicity in compare to other pluronic block copolymers and therefore results in high critical micelle concentration. 38 This is an FDA-approved polymer, and due to its long PEG tail, it can acts as steric stabilizer as well as useful in the preparation of NLCs. 39
The goal of the current work was to create a NLC system with a lipid matrix, a surfactant, and the anti-inflammatory drugs DS to enhance the drug’s release and enhance its anti-inflammatory properties. For the first time, AO was employed as the primary component in the lipid matrix in liquid lipid form, with solid lipid palmitic acid providing the crystal lattice defect that enhanced the drug loading efficiency of DS. Pluronic F-68 was also added to the formulation as a surfactant to improve the loading of DS and provide stability. The experimental setup in this work utilizes the Taguchi L9 array design to optimize and observe the effect of each element on the NLC properties. After being synthesized, the DS-loaded NLCs (DS-NLC) were added to the gel and subjected to physicochemical analysis, including pH and partition coefficient measurements, as well as analyses of PS, PDI, surface morphology, and stability studies. Moreover, the Wistar rat model was used to conduct studies on drug release, anti-inflammatory properties, and drug loading and encapsulation efficiency.
MATERIALS AND METHODS
Materials
Diclofenac sodium (DS) was received as a gift sample by Amoli Organics Pvt. Ltd., Gujarat, India; Pluronic F-68, Palmitic acid, Carbopol 934, and Almond oil (AO) were obtained from CDH Laboratories, Mumbai, India. The whole experiment was conducted using double-distilled water (DDW). The selected reagents and chemicals used in the study (including solvents and ketamine) were of analytical grade, assuring a purity level of >99%.
Methods
Optimization of drug delivery system by Taguchi experimental design L9 array
The drug-to-lipid ratio, surfactant concentration, stirring duration, and sonication time were the independent variables that were studied and their effects were statistically optimized using the Taguchi experimental design.40–42 Each variable’s level was determined to be low (1), medium (2), and high (3) (Table 1). There were three methods selected for the preparation of NLCs including solvent evaporation, solvent emulsification diffusion and micro-emulsification technique. Based on Taguchi’s design, the solvent emulsification diffusion method was found more suitable for the NLC preparation of ideal size range and other basic requirements. Therefore, solvent emulsification diffusion technique was used further for preparation of DS-loaded NLCs.
Taguchi Design L9 Array Variables
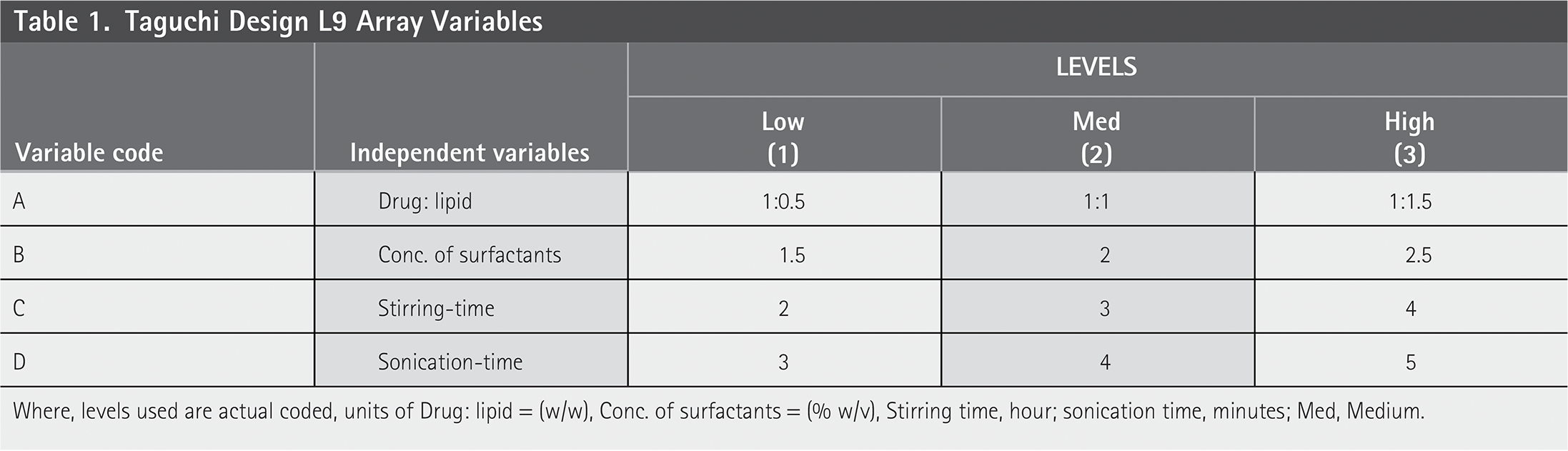
Where, levels used are actual coded, units of Drug: lipid = (w/w), Conc. of surfactants = (% w/v), Stirring time, hour; sonication time, minutes; Med, Medium.
Preparation of diclofenac sodium-loaded NLCs
Solvent emulsification diffusion was used to prepare the NLCs from the optimized data by design, as indicated in Table 1. As an internal oil phase, 150 mg of AO and accurately weighed palmitic acid were dissolved in 2 mL of a 1.4:0.6 chloroform and methanol combination. Dispersed in the aforementioned solution was 100 mg of DS. To generate an o/w emulsion, the resultant dispersion was transferred into a homogenizer tube along with 8 mL of a 1.5% (w/v) aqueous solution of the surfactant pluronic F-68 as the external aqueous phase. The homogenizer was run for 30 min at 7,514 g. Following homogenization, the aforementioned emulsion was added to a 100 mL solution of pluronic F-68 surfactant, shaken for 4 h, then centrifuged for 15 min at 22,542 g (Remi CPR-24 Plus, REMI, ELEKTROTECHNIK LIMITED, Maharashtra, India) to extract the drug-containing nanostructured lipid material. The NLCs were then obtained in pellet form by re-dispersed into 1.5% (w/v) off a water-soluble surfactant of pluronic F-68, sonicating it for 4 min at a probe sonicator (Shalom Ultrasonics, Maharashtra, India), and drying it at −80°C using a lyophilizer (Allied Frost-FD3C, New Delhi, India). Lastly, store the dehydrated mixture in the vacuum desiccator until required. 43
Designing of formulation
NLC1 through NLC9, or Nanostructured Lipid Carriers, is a group of nine NLCs that were created by combining DS, lipid matrix (which included AO, pluronic F-68, and palmitic acid), and surfactant. Following the optimized formulation, various compositions were produced by varying the ratios of (drug:lipid matrix). Table 2 provides the precise makeup of each NLC. The drug, lipid matrix, and surfactant were all weighed before being uniformly mixed and subjected to sonicate for 7 min. The produced formulations were stored in Eppendorf tubes as a pre-concentrate combination and kept at 4 ± 2°C in the refrigerator. The purpose of this storage technique was to increase the NLCs’ stability for use in later studies. To assess the anti-inflammatory activity in a gel form, all of the developed formulations—named GNLC1 through GNLC9 and listed in Table 3—were combined with carbopol 934, or a gelling agent. 44
Optimization of Variables and Characterization of Formulation Developed as per Taguchi’s Model
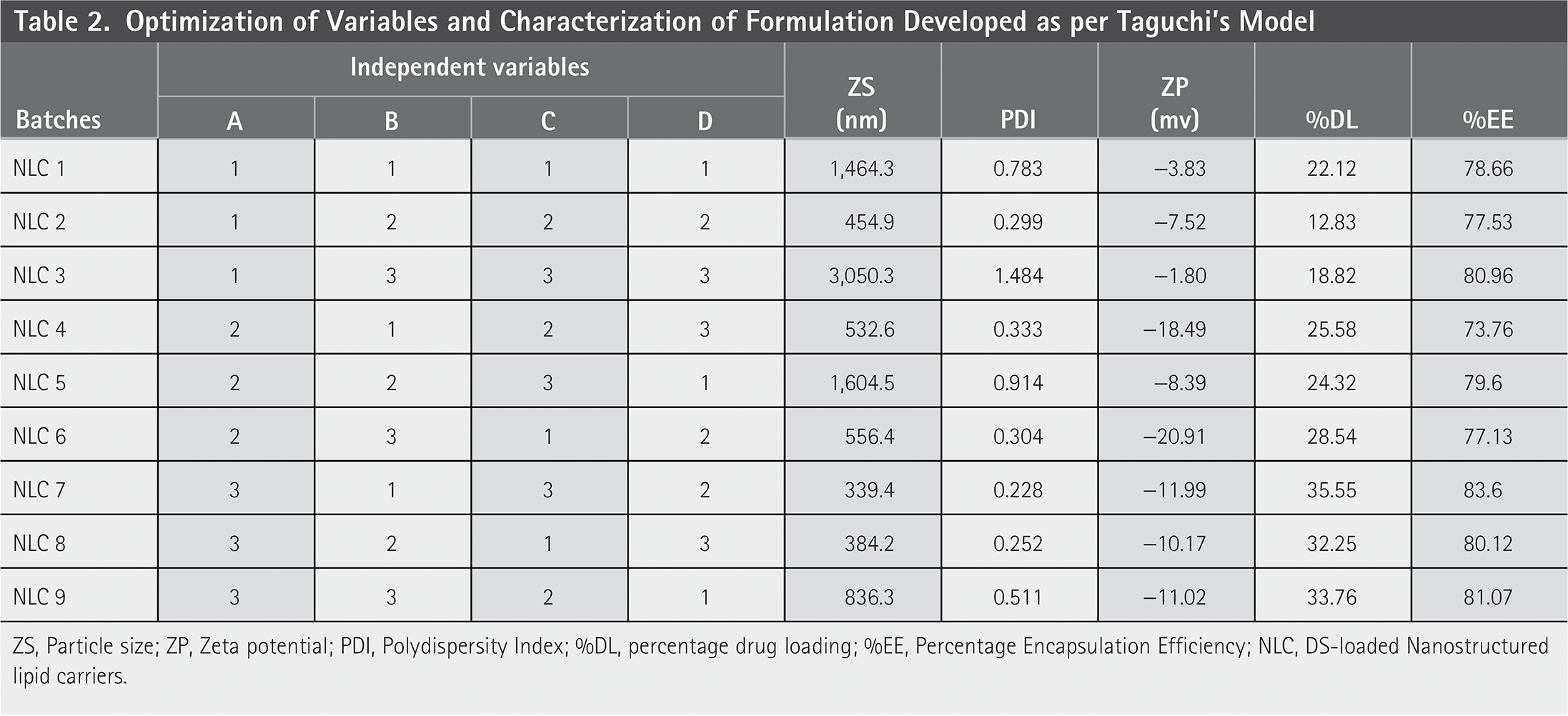
ZS, Particle size; ZP, Zeta potential; PDI, Polydispersity Index; %DL, percentage drug loading; %EE, Percentage Encapsulation Efficiency; NLC, DS-loaded Nanostructured lipid carriers.
Composition of Diclofenac Sodium-GNLCs with Different Component Proportions and Physical Property Characterization (Mean ± Standard Deviation)

Quantities in mg; Physical properties of Diclofenac sodium-loaded NLCs gel (DS-GNLC) were determined.
D:L, Drug: Lipid; Lm, Lipid matrix (palmitic acid:almond oil); Vis, Viscosity.
Physicochemical characterization
pH, partition coefficient, and viscosity measurement
Using a pH meter (EUTECH Instruments, Singapore), the diluted NLC’s pH was measured. 1 mL quantities of distilled water and n-octanol were used in this process. After that, 50 mg of the material was added to an Eppendorf tube and stirred at 25 ± 2°C for 3 h. Utilizing the UV-spectrophotometric approach (UV-Pharmaspec-1700, Shimadzu, Japan), each layer containing the drug component was identified. For DS, the evaluation was done at 282 nm, and a comparison was made with a blank that was treated identically. After that, a predetermined percentage of the sample was taken from the aqueous layer and used the following formula to quantify the partition coefficient (p).
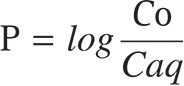
The viscosity of the GNLCs was tested in the context of the formula under nondiluted conditions using a viscometer (Model: CAP2000, Brookfield Ametek, USA), temperature of 25 ± 2°C was used for the measurements. 45
Drug loading (%DL) and drug entrapment efficiency (%EE)
The ultracentrifugation method was used to determine the % EE in NLCs. A cooling centrifuge (Remi C-24 India) was used to centrifuge several DS-NLC formulations with different lipid ratios for 50 min at 9,300 g and 25°C. The liquid supernatant, which contained unentrapped DS, was obtained and subjected to quantitative analysis using the UV technique at a wavelength of 282 nm. After comparing the %EE of each sample, the best formulation was selected for further testing.
%EE is calculated by using this equation (n = 3):

Where,
X1 = Initial amount of DS
X2 = DS quantified in supernatant
The %drug-loading (%DL) is calculated to evaluate the total amount of drug (DS) loaded into the developed system, i.e., NLC by using this equation (n = 3).39,44
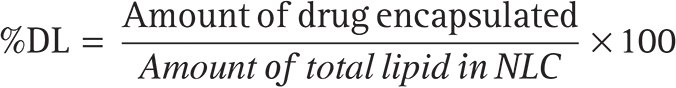
Determination of particle size, PDI, zeta potential, and morphology study
With the help of a zetasizer device (Malvern Nano ZS/ZEN-3600, USA), photon correlation spectroscopy was used to calculate the average PS and PDI of the NLCs. To avoid multiscattering, the formulation was evaluated at a concentration of 1 mg/mL and diluted 1:100 with Millipore water before size assessment. 46 To lower mistakes, every experiment was run three times.
Further verification of NLCs’ dimensions and surface features was done with transmission electron microscopy (TEM) (JEM-1400, JEOL, USA). Freshly produced DS-loaded NLCs were used at a concentration of 1 mg/mL and diluted 1:100 with double-distilled water. A 200-mesh copper grid coated with carbon and containing a single drop of the solution of the sample was used (Ted Pella Inc., USA). There were three iterations of this process. The sample was then air-dried after being treated with 2% phosphotungstic acid for negative-staining. Subsequently, the sample was subjected to a voltage increase of 120.0 kV under the TEM apparatus. 44
Stability studies
One of the parameters that must be examined in the developed drug-loaded formulations is stability. All formulation suspension solutions were made for this investigation in distilled water at a concentration of 1 mg/mL. Using a photon-correlation spectroscopy (PCS), the size, PDI, and zeta potential of DS-NLC were assessed after 1, 5, 7, 15, 30, 60, and 90 days, while temperature-condition was maintained at 25 ± 2°C. The experiments were performed in the triplicate manner. 44
Analytical methodology
Using RP-HPLC (reverse-phase high-performance liquid chromatography), an HPLC analytical instrument (Waters, 515 HPLC-Pump, India) was used to quantify the amounts of DS in permeation investigations. Component separation was performed using a Cosmosil ®C18 reversed-phase column (25 cm × 4.6 mm, 5 µm pore size; Merck, Germany), with a mobile phase made up of a 60:40:0.5 v/v/v mixture of methanol, water, and acetic acid. 0.7 mL/min was the flow rate, 5.06 min was the retention duration, and 282 nm was the detection wavelength.2,44
Ex vivo permeation studies
The male Wistar rats’ clear excised skin from the abdomen was obtained, and its hair was removed. It was then cleaned at pH 7.4 using phosphate buffer saline (PBS). Following the fat removal process using isopropyl alcohol, the skin was cleaned with distilled water and kept at −20°C temperature conditions till further requirements. All of the penetration tests were conducted on rat abdominal skin that was fully thick. The vertical Franz-diffusion cell assembly (Orchid Scientific and Innovative India Pvt. Ltd, India) with an effective diffusion area of 3.14 cm2 and a cell volume of 15 mL had the skins clamped in between the donor and receptor chamber.
A new PBS mixture was poured into the receptor chambers. By employing a recirculating water bath to keep the diffusion cells at 37°C, the fluid filled in the receptor chambers was constantly agitated at 940 g. One milliliter of every formulation, or 10 mg of the drug, was carefully inserted into the donor chambers. To maintain the sink conditions, 1.5 mL samples were taken out and replaced with PBS medium at regular intervals (0, 2, 4, 6, 8, 12, and 24 h) and then the samples were analyzed with following experiment in triplicate manner. 42
Animals and husbandry
The study used male Wistar rats, which were procured from the I.T.S College of Pharmacy, Ghaziabad, central animal house facility. At the beginning of the trial, Wistar rats weighed between 200 and 250 g, and they were kept individually in stainless-steel cages. The experimental animal room had a temperature of 20°C and a relative humidity of 50% (±5%). The lighting was adjusted to provide artificial light for 12 h every day. Standard laboratory meals were fed to the animals along with an unfettered supply of drinking water. Hair clippers and scissors were used to remove hair from the dorsal side of the Wistar rats the day before the irritation trial. To remove all of the hair from the 2 cm2 area, a professional hair remover was used. Surgical spirit was used to clean the shaved region. The test animal’s affected area was then treated with a 0.075 gm GNLC gel of DS the next day. 47
In vivo primary skin-irritancy test
One test material was selected having 1%GNLC with carbopol gel of DS which was optimized and characterized as shown in Tables 3 and 4. The dose of the test material was taken at 0.075 g (equivalent to 75 mg of DS).
48
An experimental animal’s skin receives a single dose of the chemical to be evaluated; the control animal’s untreated skin patches are used as a reference. To provide a thorough assessment of the results, the degree of irritation or corrosion is further described and evaluated at predetermined intervals. The study’s duration was adequate to assess if the changes that were noted were reversible or irreversible. Subjective evaluation was used to rate the skin reaction at the application location once every day at 1, 24, and 48 h following gel application. The Primary Dermal Irritation Index (PDII) was calculated with the help of the following formula:
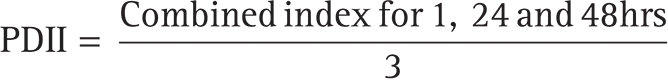
Equation and R2 of Optimized Nanostructured-Lipid Carrier and Optimized GNLC
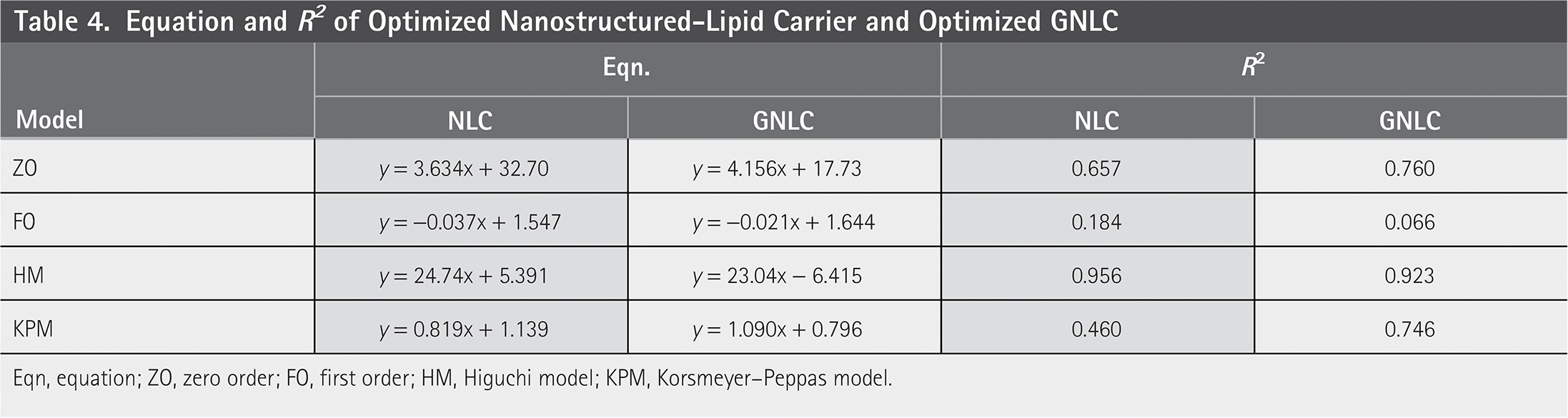
Eqn, equation; ZO, zero order; FO, first order; HM, Higuchi model; KPM, Korsmeyer–Peppas model.
Anti-inflammatory activity (carrageenan-induced paw-edema method)
A plethysmometer (MCORP, India) was used to measure changes in the volume of the inflammatory paw that resulted from injecting carrageenan (0.1 mL, 1%w/v) to assess the anti-inflammatory activity. Weighing male Wistar rats, we marked each one’s right hind paw immediately behind the tibia–tarsal junction. To guarantee constant paw volume, the paw was therefore immersed in the plethysmograph (mercury displacement method) up to the specified mark each time. Wistar rats were split up into four different groups, each consisting of six animals, plus a control group. At 0, 0.5, 1, 2, 4, 8, 24, and 48 h, the paw volume was recorded. 48
Rats were administered the formulations (GNLC) transdermally. Animals in the controlled group received injections of 0.9% NaCl saline, which was drug free. Rats were given carrageenan (in 0.9% normal saline) subcutaneously in the subplantar region of their right hind paws after 30 min of transdermally administered formulations (GNLC standard). Paw volumes were measured up to 24 h after injection, starting with the first reading immediately after. The following formula was used to determine each group’s % edema inhibition brought about by carrageenan:
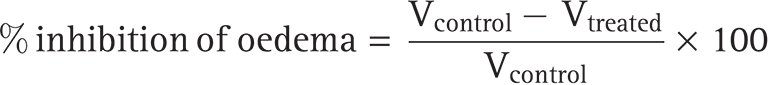
Where,
Vtreated is the edema volume of each rat in the test group and Vcontrol is the mean edema volume of the rats in the controlled group.
Statistical analysis
GraphPad-Prism (version 9.4.0) was used to do statistical analysis on the data with significant differences (p < 0.05), which are shown as mean ± standard deviation. For statistics, one-way Analysis of Variance (ANOVA) and two-way ANOVA were used as per requirements.2,47,49
RESULT AND DISCUSSION
Optimization by Taguchi Design
Based on drug loading and PS, the preparation technique was optimized. The findings of the various techniques utilized to prepare NLCs showed that the solvent emulsification diffusion technique produced the best results and prevented aggregation, hence it was chosen for additional research. It was done to optimize the drug–lipid ratio in various formulations. It was discovered that the ideal drug: lipid ratio was 1:1. Figure 1 illustrates the optimized surfactant concentration and stirring time for a drug lipid ratio formulation of 1:1. Taguchi design gives a clear emphasis on the significance of solvent emulsification diffusion technique over other methods. By varying such levels of parameters including surfactant concentration, drug and lipid ratio and the stirring time, results in the preparation of NLCs identified on the basis of their PS by DLS method. As the amount of lipid or surfactant varies it results in the larger PS which fails the development of stable NLCs. Therefore, 1:1 ratio of drug:lipid was found good for preparation of NLCs as shown in Figure 1.
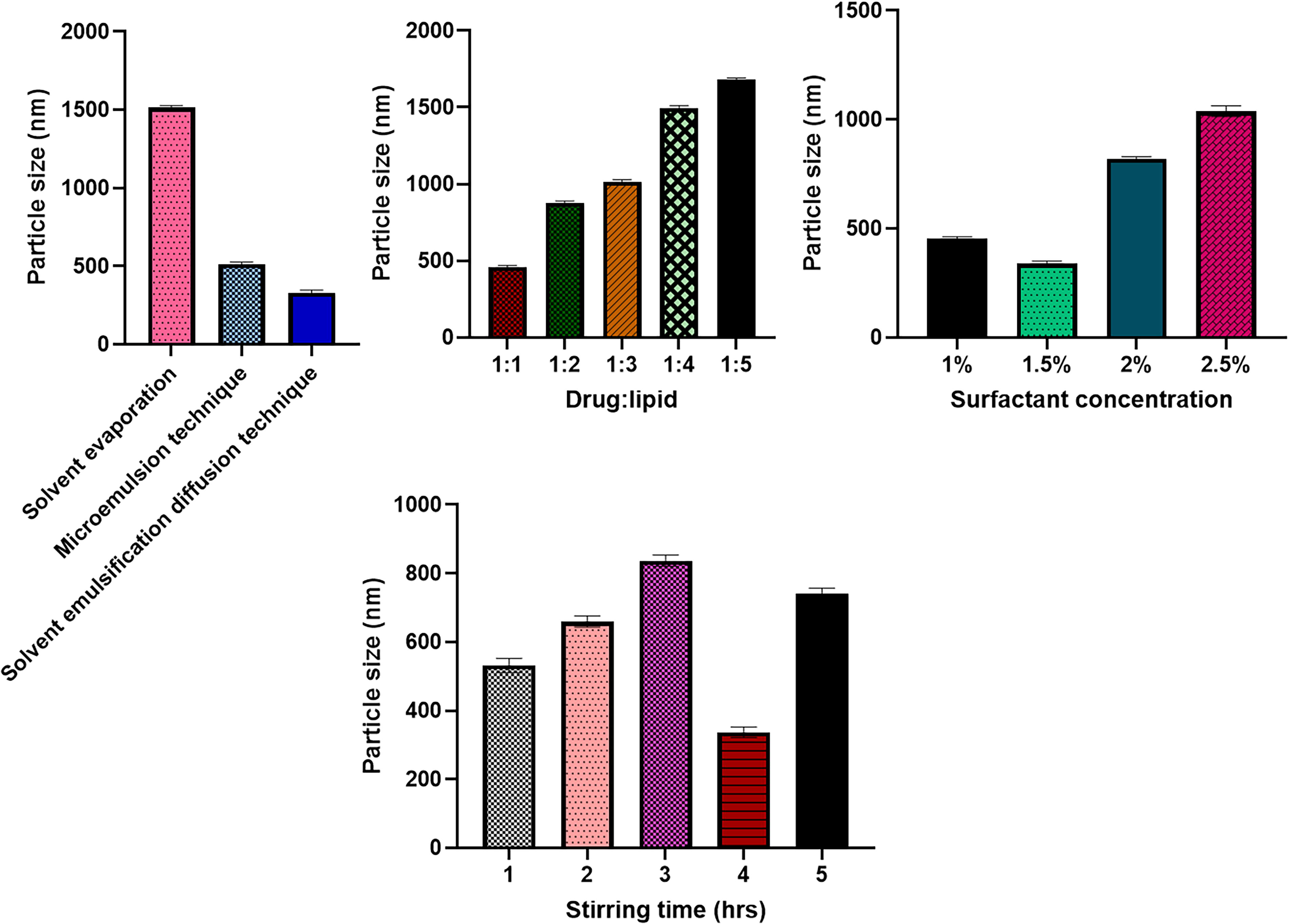
Optimization of NLC preparation method, Drug: Lipid Ratio, Surfactant-Concentration, stirring-time. NLS, nanostructured-lipid carrier.
Physical Characterization of NLCs
pH, partition coefficient, and viscosity measurement
It was found that the NLCs’ pH range was 5.47 ± 0.26. It was observed that the partition coefficients for DS and DS-NLC were 2.43 ± 0.41 and 0.343 ± 0.02, in that order. The partition coefficient moving from DS to DS-NLC demonstrates the NLCs’ solubility. It clearly indicates the higher aqueous solubility of DS-NLC. The viscosity of DS-NLC was found to be between 174 and 290 × 103 cps, which is within an acceptable range and indicates that the formulations can be useful for required purpose.
Drug-loading (%DL) and entrapment-efficiency (%EE)
Table 2 displays the results of %DL and %EE. The optimized batch, NLC7, had drug loading and drug entrapment efficiencies of 35.55% and 83.6%, respectively. %DL clearly indicates in the Figure 2 that NLC 7 has higher %DL value in compare to other NLCs in which the drug: lipid content was in higher proportion (1:1.5) with lower concentration of surfactant (1.5) by maintaining high stirring time and sonication time was set at mid-level for 4 min. These parameters were varied among other NLCs which clearly indicates that higher drug loading and lipid contents results in a good NLC preparation. Also, in NLC8 and NLC9 the lipid content was higher but there was variation in other parameters which does not show as much effect as reported previously in NLC7. Therefore, NLC7 was most suitable and better preparation for DS-loaded NLC. Similarly, %EE for NLC7 was found more in compare to other NLC formulations.
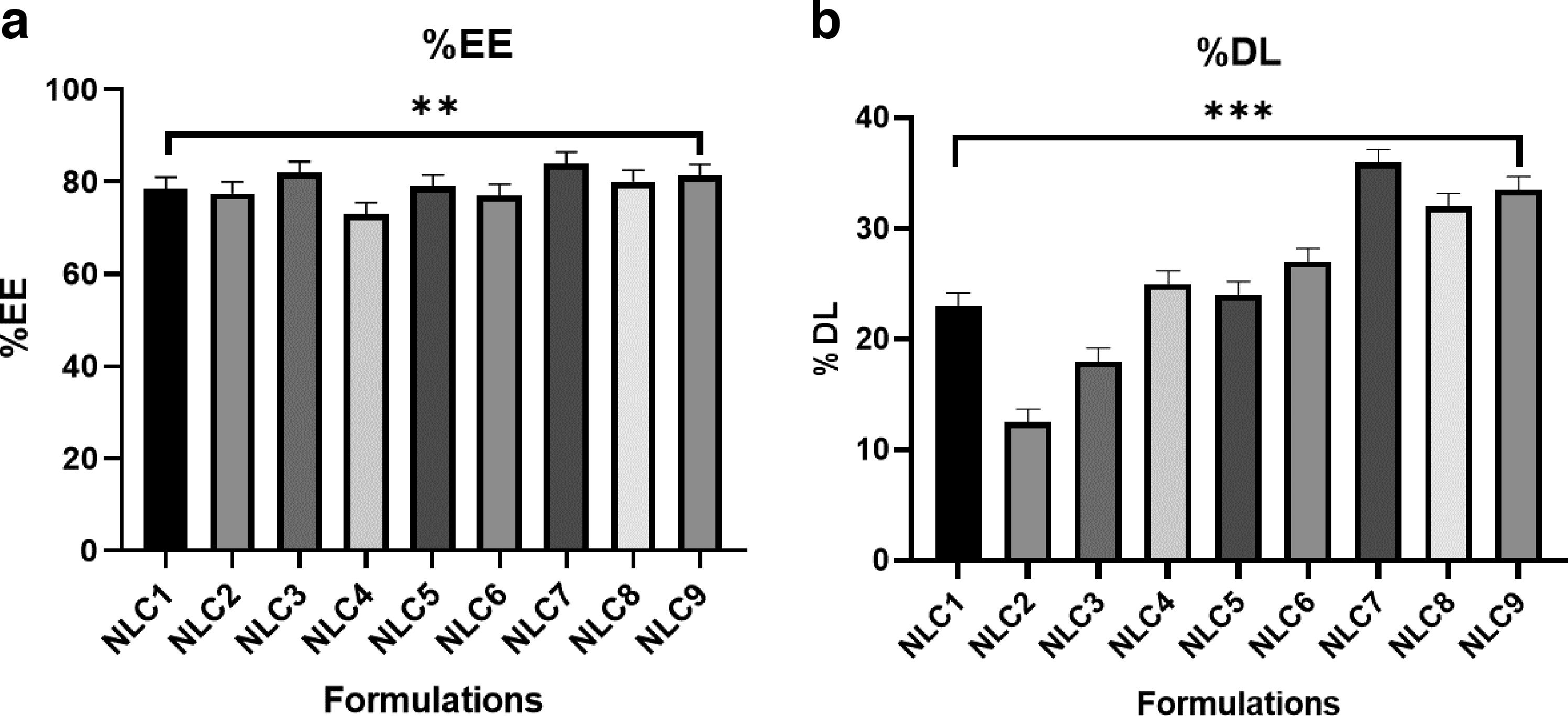
Characterization of DS-loaded NLCs
Particle size, PDI, zeta potential, and morphology of NLCs
For measuring PS, PCS is the most effective method. It gauges the dynamically scattered laser light’s intensity variation, which is brought on by particle movement. It was discovered that the optimized formulation’s PS and PDI were, respectively, 392.67 ± 58.48 nm and 0.259 ± 0.036. Due to electric repulsion, charged particles (high zeta potential) often have a lower chance of aggregating. Aggregation is facilitated by a lower zeta potential. It was discovered that the optimized NLC has a zeta potential of −9.89 ± 2.24 (Table 2) (Figure 3).
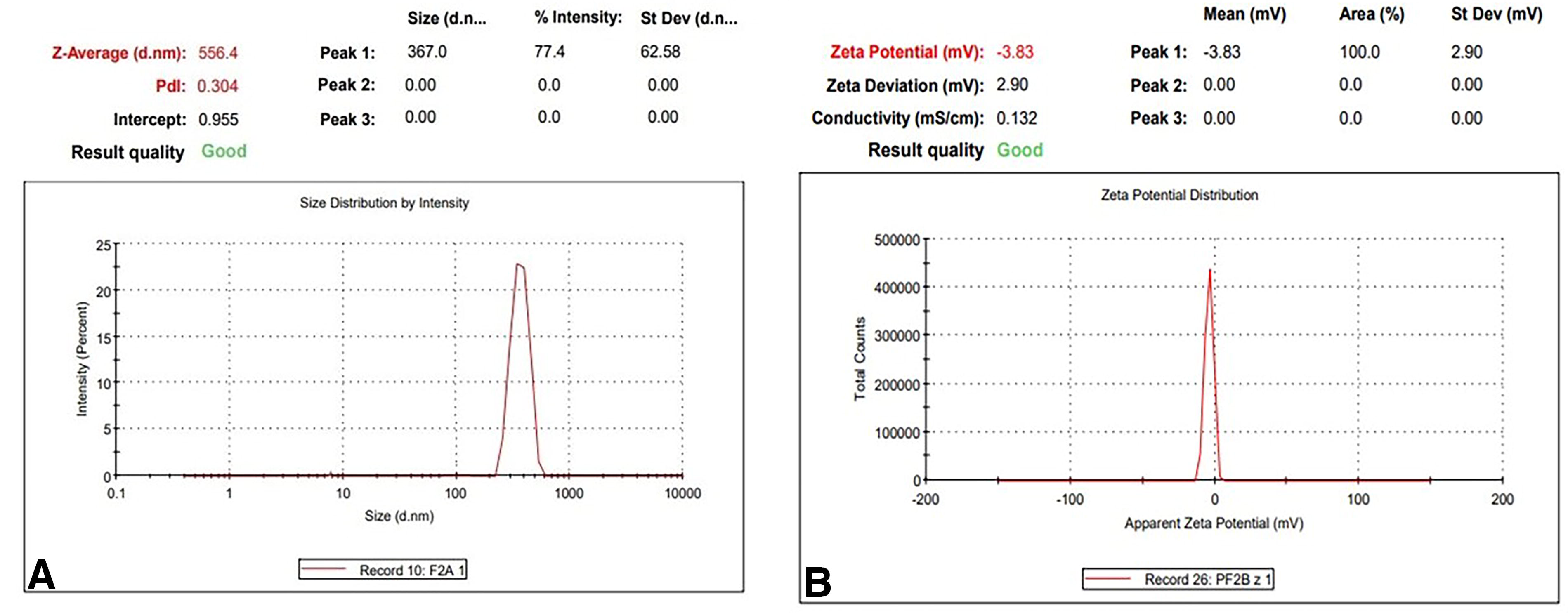
Physical characterization of NLC by photon correlation spectroscopy
TEM was used to assess the surface-morphology and shape of the prepared and optimized NLCs (NLC7). According to the study, the majority of NLCs had rounder, spherical shapes. The particles’ surfaces displayed a distinctive smoothness (Figure 4).

Morphological evaluation of optimized formulation of NLC.
Stability Studies
One important element that can affect the stability of lipid-based nanoformulations is storage time. The formulation was in this investigation stored at room temperature for 90 days during which time critical characteristics such as PS, zeta potential, and PDI were assessed. Figure 5a–c illustrates how storage time affects these factors, respectively. The acceptable size for NLC ranges from 10 to 1,000 nm in size which helps in targeted drug delivery over a particular site, the prepared NLC's size ranges <300 nm. The outcome suggests that the stability of the formulations is not significantly different (<1,000 nm) by storing them at room temperature. We can deduce that after being stored for 90 days at room temperature, this formulation will not change.
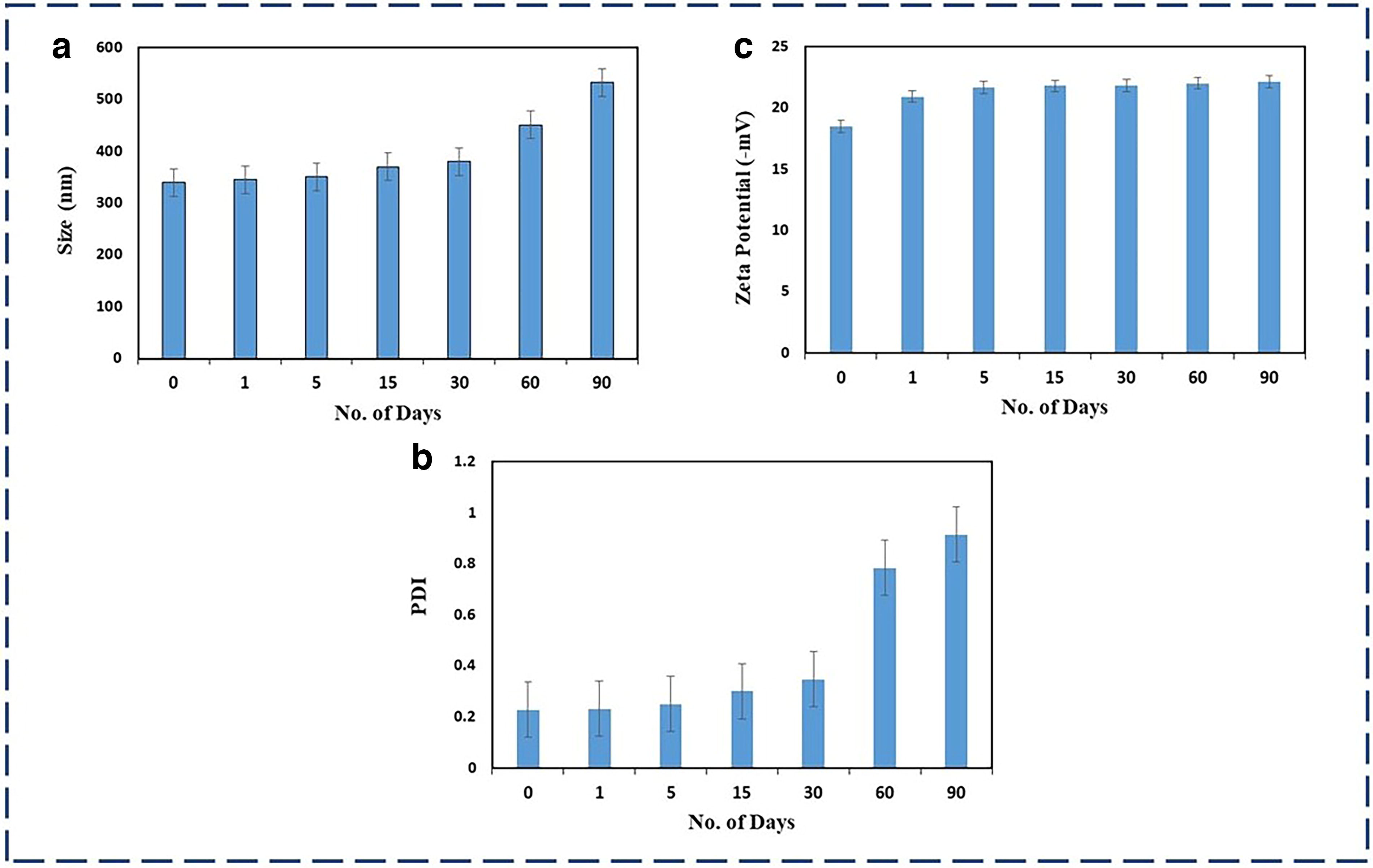
Representation of stability profile of DS-NLC by measuring the
Ex Vivo Permeation Study
Compared with pure drug formulations, the optimized NLC formulation demonstrated a significant and sustained release. Figure 6 illustrates the use of the most appropriate and prevalent formulations, NLC7 and GNLC7, which demonstrated a release of 96.34% for DS-NLC and 90.4% for gel formulation, or GNLC7, in 24 h. NLC7 has higher drug release within 24 h in compare to other NLCs which can be occurs due to higher drug: lipid content or due to variation in process parameter. There were several plots drawn to ascertain the drug-release model including cumulative %drug-release vs. time (zero-order kinetic model), log cumulative of drug-remaining vs. time (first-order kinetic model), cumulative percentage of drug-release vs. square root of time (Higuchi model), and log-cumulative of drug-release vs. log-time (Korsmeyer model). Table 4 lists the outcomes of the additional research that was conducted using the acquired equations.

%drug-release of DS permeated in an ex vivo model of rat abdominal skin from NLC and GNLC in PB at pH 7.4 with significant differences (p < 0.01) (n = 6).
In Vivo Skin Irritation Test
As a result of chemicals interacting with the skin’s sensory receptors at the application site, primary skin irritation is the development of reversible inflammatory changes in the skin after the application of a test material. Skin reaction grading ranged from no irritation to severe irritation (including both erythema and edema). The mean PDII of animals treated with standard gel was 1.49, indicating slight irritation; however, no irritation was observed when the animals were treated with DS-NLC, or prepared gel formulation, i.e., GNLC; this resulted in good gel properties as a nonirritating agent that can be used for future research.
Anti-Inflammatory Activity
Using the carrageenan approach, the anti-inflammatory potential of DS was discovered. Three groups were taken of which two groups including group 2 and group 3 were treated with standard DS gel and an optimized formulation of DS-NLC-loaded gel, or GNLC, respectively. The findings were significantly different from the standard and control groups. When the standard and test formulations were compared, the test formulations produced a more dominant inhibition of edema, which in turn produced a higher anti-inflammatory activity of the DS-NLC-based gel. The results are displayed in Figures 7 and 8. The control group was remained untreated with any pharmacological treatment.

Macroscopic images of comparison between rat paw edema in different groups

Anti-inflammatory activity evaluation of prepared formulation of DS-loaded NLC-based gel, i.e., GNLC as test formulation vs. standard DS gel
DISCUSSION
NLCs were reported as the ideal delivery system for biopharmaceutical classification system (BCS) class drugs resulting various merits over other DDS including sustained release, bioavailability enhancement and targeting delivery sometimes.44,50 The development of NLC loaded with DS, also known as DS-NLC or NLC, is described in depth in the current research. The current strategy aims to maintain drug release, adjust the drug molecule’s overall function, and modulate its anti-inflammatory effect when applied topically. Using a solvent emulsification technique, the DS-NLC formulation was developed, 42 with AO acting as the lipid matrix and DS attached to palmitic acid. Drug loading in NLC is significantly influenced by pluronic F-68; in the meantime, %drug release and %DL were shown to be significantly impacted by the lipid/drug ratio. The function of surfactants in NLC design is to emulsify the components of the oil phase and solidify the lipid matrix. The DS-NLC technology outperformed the usual gel in permeability trials carried out with the excised skin approach. 48
PS, PDI, and %EE were among the NLC properties that met acceptable standards. The higher lipid content in the optimized formulations helps in the enhancement of stability of NLCs, as the presence of pluronic F-68 modulates stability.38,39 Evaluations of stability showed that the formulation was stable for 90 days at room temperature. It clearly indicates the positive effect of formulation in the systemic circulation or at the site of action. The lipid matrix of the medication in NLC is responsible for the delayed release pattern observed in the ex vivo DS release profile of NLC when compared with naïve DS. The prolonged release seen at pH 7.4 points to the possibility of regulated drug release. Ex vivo studies also demonstrated how the binding of drug-lipids and its subsequent solidification as a gel with appropriate rheological parameters, which increases its viscosity and facilitates transport through the skin, alters drug permeation in DS-NLCs-loaded gel, or GNLC. Also, the GNLC does not show any irritation across the skin which makes it a good candidate for anti-inflammatory activity. It helps in enhancing the easy delivery of drug at affected site with good patient compliance, least adverse effects and modulated potential.
A poorly soluble drug component called DS is incorporated into an NLC system and loaded into the DS-NLC carrier system. The physical stability of NLC is indicated by its negative zeta potential. It should be mentioned that at 4°C, storage stability persisted for up to 3 months. The lipid matrix used in the system may explain the physical and storage stability of the DS-NLC. AO, pluronic F-68, and palmitic acid are the main lipid constituents. According to the preliminary assessment, NLC made of palmitic acid could prolong the drug’s release; however, NLC becomes more soluble when AO is added. In comparison to the DS-NLC system, the naive DS has a greater partition coefficient. It denoted the higher aqueous solubility of developed DS-loaded NLC in compare to naïve DS.
In contrast to the standard drugs, DS-NLC exhibits sustained release for up to 24 h. Significant heterogeneity in release pattern is produced by the lipid/drug ratio. Since DS are lodged in the lipid matrix, the hydrophilic drug that is on the vesicle’s surface can be released slowly without diffusing out of the matrix for up to 24 to 48 h. The release of the medication from the matrix in which it is embedded into lipophilic cores would be further delayed by the addition of palmitic acid.
The FDA-mandated daily limits for the nontoxic, biocompatible components used in the manufacturing of NLC were met. In addition to another aspect, drug permeability is reliant on pH and concentration gradients. Research revealed that DS-NLC permits more drug permeation than GNLC. Drug penetration may be modulated by the involvement of NLC components (lipophilic components). 44 The evidence suggesting that GNLC has a nonirritating behavior and is therefore employed for its anti-inflammatory effect was enhanced by the in vivo skin irritation test. Compared with the regular DS gel, anti-inflammatory activity in the Wistar-rats model demonstrates more substantial outcomes. Adding them to NLC and loading them as a gel form for transdermal distribution, demonstrates the improvement in the anti-inflammatory potential of DS.
CONCLUSION
The goal of the study is to solve the issues brought on by the BCS Class II drug’s (DS) poor solubility and constrained skin release. The process of designing and development of NLCs containing DS, also known as DS-NLC or NLC. The physical characterization comprised pH, %DL, %EE, and partition coefficient measurement. Different combinations of solid lipids (palmitic acid), liquid lipids (AO), and the surfactant pluronic F-68 were used to produce DS-loaded NLCs. The anti-inflammatory efficacy, size, and drug release of the formulations were assessed. NLCs were effectively generated in a nanostructure, according to TEM imaging and stability data. When DS-NLC was diluted, the droplets showed diameters <1,000 nm. Ex vivo drug release investigations using a rat skin model showed increased permeability of drug-soluble solids (DS) from DS-NLC in comparison to pure drugs, possibly because of NLC production. According to the study’s findings, developing a delivery system based on DS-NLC improves the solubilization of DS-NLC, increases the assimilability of DS, and amplifies the anti-inflammatory effect of DS-loaded gel, or GNLC. Ex vivo release studies conducted at pH 7.4 conditions showed that DS-NLCs sustained DS release. Since the lipid nanocarrier system integrated into GNLC gel form did not cause skin irritation, it was a good option for anti-inflammatory activity. Due to AO’s improved nanocore-formation, the study showed that NLCs loaded as gel, or GNLCs, had a strong anti-inflammatory potential when compared with ordinary DS gel. The developed NLCs for DS results as a promising and effective DDS from the aspect of pharmaceutical industry.
IAEC ETHICS APPROVAL
Study design conducted on animals was approved (ITS/06/IAEC/2018) by Institutional Animal Ethics Committee (IAEC) of ITS, Ghaziabad, 201206, Uttar Pradesh, India.
AUTHORS’ CONTRIBUTIONS
A.P.S., I.C., and M.V. were responsible for conceptualization, project designing, and experimentation. D.P. and J.K. validated the work. M.Y. supervised the work. P.D. and S.A. wrote the initial draft with the help of N.K. A.J. wrote and reviewed the article. A.P.S. finalized the article. P.K.S. and N.K. helped in data curation.
Footnotes
FUNDING INFORMATION
No funding was received for this article.
DATA AVAILABILITY STATEMENT
Required data is already attached in the article and there are not any supplementary data to show.
DISCLOSURE STATEMENT
No competing financial interests exist.