
Abstract
Little is known about the HIV-1 epidemic in Balkan countries. To fill the gap, we investigated the viral genetic diversity in Bulgaria, by sequencing and phylogenetic characterization of 86 plasma samples collected between 2002 and 2006 from seropositive individuals diagnosed within 1986–2006. Analysis of pol gene sequences assigned 51% of the samples to HIV-1 subtype B and 27% to subtype A1. HIV-1 subtype C, F, G, H, and a few putative recombinant forms were also found. Phylogenetic and molecular clock analysis showed a continuous exchange of subtype A and B between Bulgaria and Western as well as other Eastern European countries. At least three separate introductions of HIV-1 subtype A and four of HIV-1 subtype B have occurred within the past 25 years in Bulgaria. The central geographic location of Bulgaria, the substantial genetic heterogeneity of the epidemic with multiple subtypes, and the significant viral flow observed to and from the Balkan countries have the potential to modify the current HIV-1 epidemiological structure in Europe and highlight the importance of more extensive and continuous monitoring of the epidemic in the Balkans.
Introduction
C
According to recent serological surveys conducted in Bulgaria's largest cities and towns, the prevalence of HIV-1 infection is only 0.73% among commercial sex workers and 0.59% among injecting drug users, whereas no infection has been found among the Roma population. 2 However, the high incidence of HCV infection and syphilis and the high level of risk behavior in these population groups suggest that they are quite vulnerable to HIV infection and thus represent a potential source of infection. 3 –5
Following the end of the cold war a decade ago, most Balkan countries have been affected by dramatic political and socioeconomic changes, which could contribute to the spread of HIV infection. Given the central geographic location of Bulgaria at the crossing point between Western Europe, Eastern Europe, and the Middle East, defining the origin and evolution of HIV in Bulgaria has obvious epidemiological relevance. However, although basic information on the characteristics of HIV-1-infected individuals in Bulgaria is available, little is known about the HIV origin and distribution of different circulating subtypes. The small amount of information from other Balkan countries indicates that there is high genetic diversity, with subtype B being predominant in Yugoslavia and subtype A in Albania. 6,7
The objective of the present study was to investigate the molecular diversity and epidemiology of HIV-1 subtypes circulating in Bulgaria. To this end, we sequenced a fragment of the HIV-1 pol gene amplified from serum samples collected between 2002 and 2006 within a cohort of infected subjects under monitoring in different Bulgarian hospitals. By employing high-resolution phylogenetic and phylogeographic analysis, as well as molecular clock estimates, we obtained, for the first time, detailed information about the origin and the epidemiological history of the two most prevalent viral subtypes (HIV-1 A and B) in this Balkan country.
Materials and Methods
Patient samples
Eighty-six serum samples were obtained from persons who were diagnosed with HIV infection [58 treated with highly active antiretroviral therapy (HAART), 23 naive, and 5 no treatment data available] between 1986 and 2006 in Bulgaria. The serum samples were obtained from EDTA-whole blood (centrifugation at 3000 rpm at 18°C for 15 min), and stored at −80°C at the National HIV Confirmatory Laboratory in Sofia prior to genotype analysis. The samples were linked to demographic and clinical data through an anonymous numerical code, in accordance with the ethical standards of the National HIV Confirmatory Laboratory of Sofia. We successfully performed HIV-1 genotyping on 83 (97%) of these samples (56 males, 27 females).
Pol gene sequencing
Sequencing and genotyping of the protease (PR) and reverse transcriptase (RT) of HIV-1 pol gene were performed in the National HIV laboratory in Sofia, Bulgaria by a commercially available diagnostic test. PR and RT pol sequences were generated after RNA extraction, reverse transcriptase polymerase chain reaction (RT-PCR) amplification, and automatic DNA sequencing, performed with the Applied Biosystems Viroseq HIV-1 Genotyping System (Abbott Wiesbaden Germany) following the manufacturer's instructions. Viral RNA extraction was performed using a Roche Amplicor HIV-1 Monitor with an Ultrasensitive Specimen Preparation kit (alcohol extraction). The PCR products (1.8-kb amplicons) were sequenced using seven different overlapping sequence-specific primers, capillary electrophoresis, and laser detection; the sequencing samples were analyzed with the ABI Prism—310 Genetic Analyzer using Viroseq analysis software (Applied Biosystems, Foster City, CA).
Genetic subtyping and phylogenetic analysis
All 83 HIV-1 pol sequences were first analyzed using the REGA HIV-1 Subtyping Tool.
8
The sequences were aligned using ClustalX software
9
followed by manual editing using the Bioedit software.
10
The final data sets included HIV-1 pol Bulgarian strains, as well as subtype-specific and circulating recombinant form (CRF) reference sequences downloaded from the HIV Los Alamos database (
Statistical support for specific clades in each phylogeny was obtained with the ML-based zero branch length test for the ML tree, 12 by bootstrapping (1000 replicates) for the NJ trees, and by calculating the posterior probability of each monophyletic clade for the Bayesian tree. Trees were rooted by ML rooting by selecting the rooted tree with the best likelihood under the molecular clock constraint or by outgroup rooting. The nucleotide sequences obtained in this study have been submitted to GenBank under accession numbers EF517409–EF517489. Multiple sequence alignments of Bulgarian and reference HIV strains are available from the authors upon request.
Molecular clock analysis
Since both subtype A and subtype B strains (the two main subtypes responsible for about 75% of the infections in our sample, see Results) were collected at different times within a 4-year period, it was possible to use their different sampling dates to calibrate a molecular clock. We assembled two separate data sets for HIV-1A and HIV-1B sequences, each one including Bulgarian strains, as well as a subset of those reference sequences for which sampling dates were available in the Los Alamos HIV database. Ultrametric trees were obtained by enforcing a molecular clock on the inferred genealogy and reestimating the branch lengths and substitution parameters with ML using the previously selected evolutionary model. The clock hypothesis was tested with the likelihood ratio test. Evolutionary rates and divergence times for the major Bulgarian clades in the HIV-1 subtype A and B phylogenies were also estimated with the Bayesian framework implemented in the BEAST program using the HKY+, and a relaxed molecular clock model that employed an exponential distribution of evolutionary rates as before.
13
For each alignment, three parallel MCMC were run and combined with the program LogCombiner v1.4 [
Phylogeographic analysis
The hypothesis of restricted gene flow among distinct HIV-1 populations in Bulgaria and other Eastern and Western European countries was tested with a modified version of the Slatkin and Maddison test 14 that counts migration to/from different viral subpopulations, 15 using the MacClade version 4 program (Sinauer Associates, Sunderland, MA). A one-character data matrix is obtained from the original data set by assigning to each taxon in the tree a one-letter code indicating its country (or geographic region) of origin. Then, the putative origin of each ancestral sequence (i.e., internal node) in the tree is inferred by finding the most parsimonious reconstruction (MPR) of the ancestral character. The final tree length, i.e., the number of observed migrations in the genealogy, can easily be computed and compared to the tree-length distribution of 10,000 trees obtained by random joining-splitting. Observed genealogies significantly shorter than random trees indicate the presence of subdivided populations with restricted gene flow. Specific migrations among different countries (character states) were traced with the State changes and stasis tool (MacClade software), which counts the number of changes in a tree for each pairwise character state. When multiple MPRs were present (as in our data sets), the algorithm calculated the average migration count over all possible MPRs for each pair. The resulting pairwise migration matrix was then normalized, and a randomization test with 10,000 matrices obtained from 10,000 random trees (by random joining-splitting of the original tree) was performed to assess the statistical significance of the observed migration counts.
Results
Diversity of HIV-1 subtypes circulating in Bulgaria
Of the 83 pol gene sequences, 41 (51%) were classified as subtype B, 18 (22%) as subtype A1, 6 (7%) as subtype C, 3 (3.7%) as subtype F, and 2 (2.5% each) as subtype H or G, respectively. The subtype analysis also classified eight sequences (10%) as putative CRFs (five as 01_AE, one as 02_AG, and two as 05_DF), whereas the remaining three (3.5%) sequences could not be classified into known groups (data not shown). The distribution of the different subtypes by age, gender, HIV exposure category, presumed place of infection, and year of HIV/AIDS diagnosis is reported in Table 1. Subtype A represented 27% of all subtypes identified after 1995, but was detected in only one individual diagnosed up to 1995, suggesting a later introduction of HIV-1A in Bulgaria. After 1995, the detection frequency of subtype A versus subtype B did not significantly change over time (χ 2 for trend >0.05).
High-resolution phylogenetic analysis of HIV-1 subtype B in Bulgaria
The ML pol gene tree of Bulgarian and other available HIV-1B reference sequences is shown in Fig. 1. NJ and Bayesian trees gave similar topologies (data not shown). The Bulgarian subtype B strains were highly interspersed within the tree. Four monophyletic clades of Bulgarian sequences, highlighted by the boxes in Fig. 1, were statistically supported in the NJ (bootstrap values >75%), ML (zero branch length test p-value <0.001), and Bayesian tree (posterior probability >90%). All four clades clustered subtype B sequences from individuals infected through sexual transmission. Sequences from individuals infected through other transmission routes were intermixed in the tree and failed to cluster within any supported monophyletic clade. Bulgarian subtype B clades I and III appeared to be more closely related to HIV subtype B sequences from Western Europe, whereas II and IV clades from Bulgaria clustered with HIV-1 sequences from the Former Soviet Union (FSU). Clade IV in particular was closely related to strains from Ukraine. The tree also showed a clustering between two Bulgarian strains with two strains from the FSU (Russia and Bielorussia), that was not statistically supported (bootstrap <50%). The high degree of intermixing between Bulgarian and foreign sequences and the phylogenetic position of the four statistically supported clades are evidence of several independent introductions of HIV-1 subtype B into Bulgaria from both East and West Europe. Moreover, the existence of independent monophyletic clades implies the presence of separate transmission networks for subtype B within the Balkan countries.
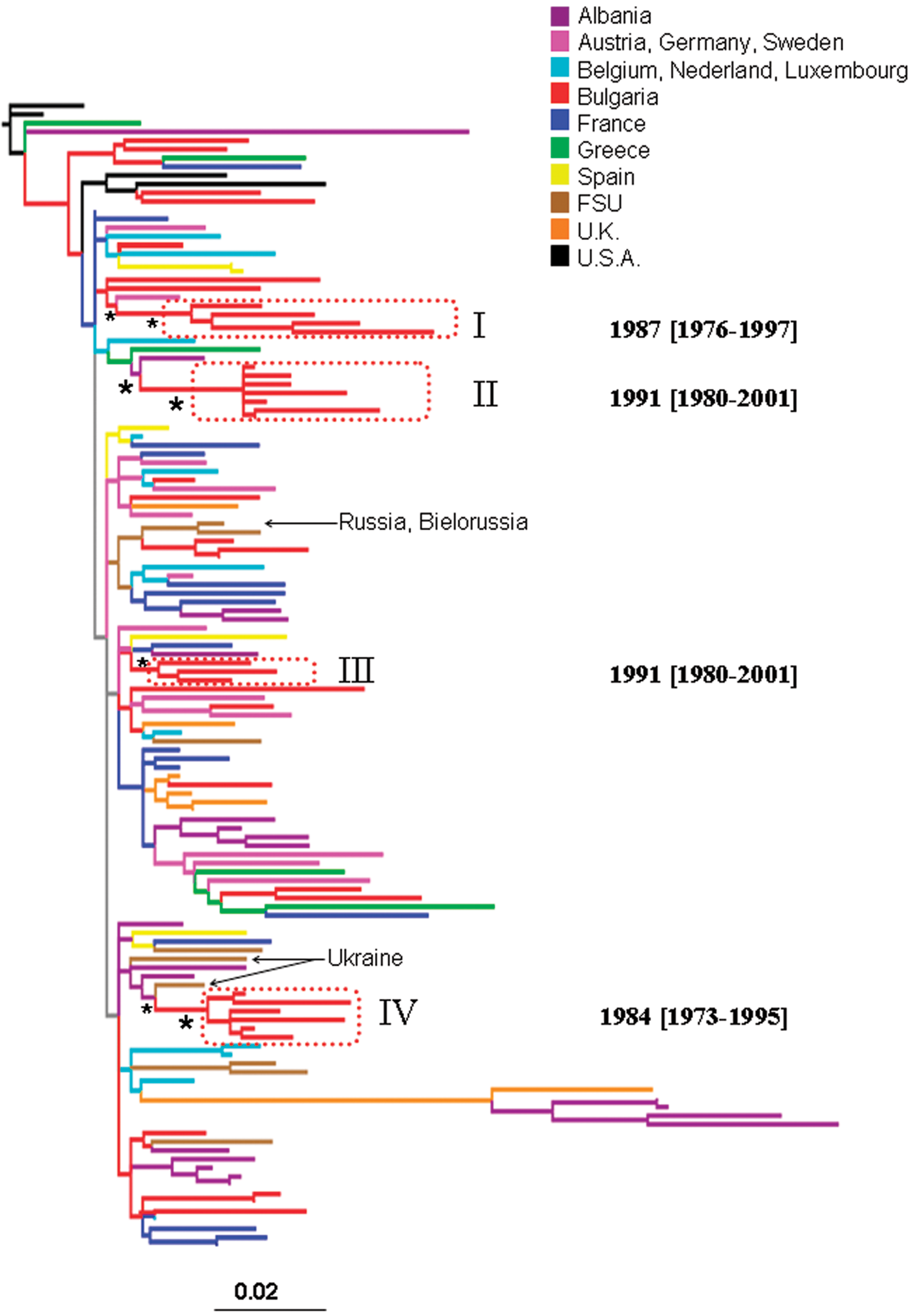
Maximum likelihood phylogenetic analysis of HIV-1B pol sequences. The data set included 41 HIV-1B strains from Bulgaria and 91 subtype B reference sequences downloaded from the Los Alamos HIV database. The tree was rooted by using two HIV-1A strains as the outgroup. Branch lengths were estimated with the best fitting nucleotide substitution model according to a hierarchical likelihood ratio test, 11 and were drawn in scale with the bar at the bottom indicating 0.02 nucleotide substitutions per site. One asterisk (*) along a branch represents significant statistical support for the clade subtending that branch (p < 0.001 in the zero-branch-length test, Bayesian posterior probability >90%, and bootstrap support >75%). The color of each external branch represents the country of origin of the sequence corresponding to that branch, according to the legend in the figure. FSU (Former Soviet Union) strains include sequences from Russia, Bielorussia, Ukraine, and Slovenia (the specific country of origin is indicated by the arrows). Broken boxes highlight statistically supported monophyletic clades including Bulgarian strains. The inferred time of the most recent common ancestor of each supported Bulgarian clade is also indicated on the right of the highlighted clade. Dates and 95% high posterior density interval (within square brackets) were estimated using a Bayesian relaxed molecular clock (see Materials and Methods).
High-resolution phylogenetic analysis of HIV-1 subtype A1 in Bulgaria
Figure 2 shows the maximum ML pol gene tree of Bulgarian and other available HIV-1A1 reference sequences. NJ and Bayesian trees gave similar topologies (data not shown). The Bulgarian subtype A strains are distributed within three statistically supported monophyletic clades (bootstrap values >75%, zero branch length test p-value <0.001, Bayesian posterior probability >90%). Strains 21A and 172A, which clustered together at the top of the tree, were isolated from two subjects infected through blood transfusion, whereas the strains in the remaining two clades (indicated in the figure as Bulgaria West Europe clade and FSU clade, respectively) were isolated from persons infected through sexual contact. The Bulgarian blood transfusion subtype A1 clade appears to be closely related to a monophyletic clade that includes both West European and African sequences. A second statistically supported monophyletic clade included most of the Bulgarian subtype A1 strains (77.8%) and HIV-1 subtype A1 sequences from Western Europe, including France and Spain. Although a monophyletic group joining a strain from the former Yugoslavia and one from Greece appeared at the origin of the clade (see Fig. 2), the clustering was not significantly supported in the ML tree and was not confirmed by the NJ or Bayesian trees. The presence of HIV-1A sequences from Africa (Cameroon), close to the root of the clade in the ML, NJ, and Bayesian trees (data not shown), with Western European strains branching off suggests an early introduction of subtype A1 from Africa to West Europe. The third statistically supported clade included two Bulgarian subtype A strains, in addition to sequences originating from different Eastern European countries. Overall, each cluster is likely to represent a separate introduction into Bulgaria of HIV-1A1 from at least two different geographic areas (West and East Europe) by different transmission routes (sexual transmission and blood transfusion).
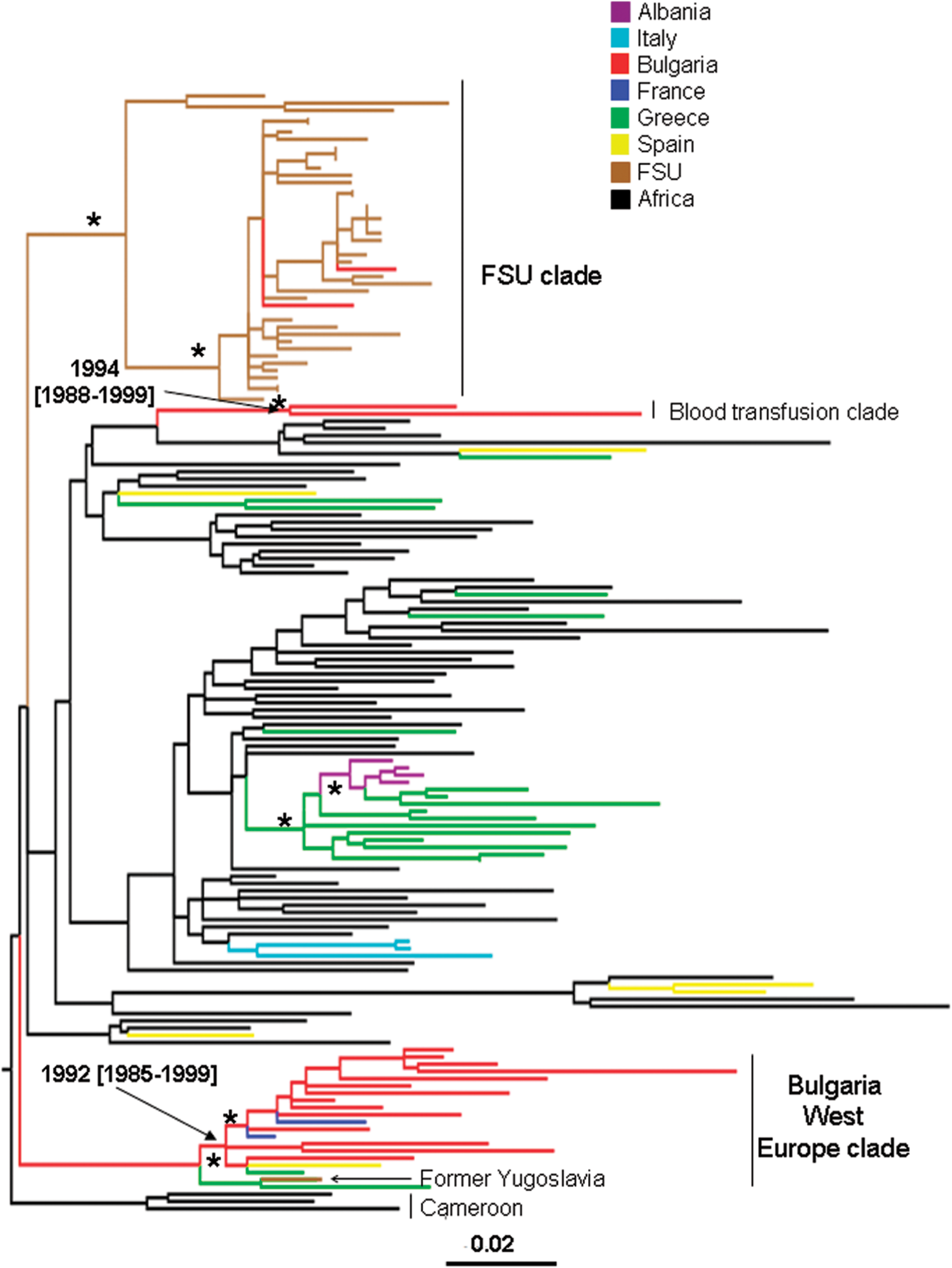
Maximum likelihood phylogenetic analysis of HIV-1A1 pol sequences. The data set included 18 HIV-1A strains from Bulgaria and 137 subtype A1 reference sequences downloaded from the Los Alamos HIV database. The tree was rooted by using two HIV-1B strains as the outgroup. Branch lengths were estimated with the best fitting nucleotide substitution model according to a hierarchical likelihood ratio test, 11 and were drawn to scale with the bar at the bottom indicating 0.01 nucleotide substitutions per site. One asterisk (*) along a branch represents significant statistical support for the clade subtending that branch (p < 0.001 in the zero-branch-length test, Bayesian posterior probability >90%, and bootstrap support >75%). The color of each external branch represents the country of origin of the sequence corresponding to that branch, according to the legend in the figure. The Blood Transfusion clade and Bulgaria West Europe clade are the two major monophyletic clades containing the Bulgarian isolates. The FSU (Former Soviet Union) clade includes strains from Russia, Bielorussia, Ukraine, Slovenia, Czech Republic, and two strains from Bulgaria. Dates and 95% high posterior density intervals (within square brackets) were estimated using a Bayesian relaxed molecular clock (see Materials and Methods).
Molecular clock analysis of the major HIV-1 Bulgarian clades
To estimate the time of the most recent common ancestor (tMRCA) of the supported Bulgarian clades within subtype A and B, we implemented a strict molecular clock model, assuming a constant evolutionary rate along each subtype tree. Since the molecular clock hypothesis was rejected for both subtype A and subtype B isolates (p < 0.0001), we calibrated the rate and the origin of the different HIV-1 Bulgarian clades by relaxing the strict clock model and allowing the evolutionary rate to vary along the tree by sampling branch-specific rates from an exponential distribution (see Materials and Methods). The tMRCA for the different clades within subtype B and A are shown in the trees in Figs. 1 and 2, respectively. It was not possible to date the introduction of HIV-1A in Bulgaria from Eastern Europe (FSU clade in Fig. 2) because only two Bulgarian strains intermixed in the FSU clade were available. The other two subtype A Bulgarian clades (Fig. 2), as well as clade II and III within subtype B (Fig. 1), originated in the early 1990s, while the origin of subtype B clade I and III was estimated to be in the mid-1980s (Fig. 1).
Migration patterns of HIV-1 A1 and B subtypes to/from Bulgaria
The trees given in Figs. 1 and 2 were also analyzed with a modified version of the Slatkin and Maddison method 14,15 to infer the HIV-1 gene flow (migration) between Bulgaria and other geographic areas. To perform the analysis, the HIV-1 non-Bulgarian sequences were assigned to three distinct groups: Africa, West Europe, and East Europe for subtype A1, or the United States, West Europe, and East Europe for subtype B. The null hypothesis of panmixia (i.e., no population subdivision or complete intermixing of sequences from different geographic areas) was rejected by the randomization test (p < 0.0001) for both subtypes. 14 Almost 19% of the observed HIV-1 subtype A1 gene flow to Bulgaria was from Africa and East and West Europe (Fig. 3A). 16,17 In contrast, the HIV-1A1 gene flow from Bulgaria to West and East European countries accounted for only 5.4% of the observed migrations, which was the same order of magnitude as the migrations observed between East European and West European groups (Fig. 3A). A substantial gene outflow of HIV-1A1 strains from Africa to the other regions, including Bulgaria, was also evident (71.9% of the observed migrations, Fig. 3A). Within the HIV-1 subtype B tree, the gene flow to Bulgaria from the United States, as well as West and East European countries, accounted for 45.2% of the observed migrations (Fig. 3B). The gene flow from Bulgaria to West Europe was 5.4%, slightly lower than the gene flow from other East European countries to West Europe (8%). On the other hand, the HIV-1B gene flow from West European countries to Bulgaria and to other Eastern European countries accounted for 34.2% and 33.8% of the observed migrations, respectively. The proportion of gene flow to/from the United States displayed in Fig. 3B was probably underestimated because HIV-1B migration analysis was focused primarily on the relationship between the epidemic in Bulgaria and other European countries and included only a few U.S. isolates for comparison. 18 –26

Maximum parsimony migration patterns of Bulgarian sequences to/from different geographic areas. The bubblegram shows the frequency of gene flow (migrations) to/from different geographic areas, as the percentage of the total observed migrations estimated from the maximum likelihood trees for different subtypes with a modified version of the Slatkin and Maddison test (see Materials and Methods). The surface of each circle is proportional to the percentage of observed migrations given within the circle. The highlighted row indicates inferred HIV-1 outflow from Bulgaria to different geographic areas. The highlighted column indicates inferred HIV-1 inflow to Bulgaria from different geographic areas. (
Discussion
The present study is the first detailed investigation of the HIV-1 molecular epidemiology in Bulgaria. Although AIDS and HIV surveillance data suggest that Bulgaria, like other countries in Central Europe, still has a low AIDS/HIV incidence, data on other sexually transmitted infections indicate a potential for spread of HIV infection. For example, 21.5% of commercial sex workers, 6.7% of the Roma community, and 2.4% of intravenous drug users were found positive for syphilis. 2,3 Our findings suggest that HIV-1 infection has been present in Bulgaria for at least the past two decades and established three main characteristics of the epidemic: first, a high genetic heterogeneity in terms of circulating subtypes; second, several independent transmission clusters within subtype A1 and B (the two most represented subtypes in the screened sample), indicating multiple introductions over time but only limited introduction from countries of the FSU; and third, a dynamic HIV-1 gene inflow and outflow from/to West Europe and other East European countries.
Results of the screened population sample, which included about 15% of all known diagnosed HIV subjects in this Balkan country, clearly showed a wide genetic diversity of the circulating virus. Although subtype B was the most prevalent subtype and was identified in about 50% of the screened samples, subtype A1 was identified in approximately 25% of samples, while non-A/non-B subtypes or several putative CRFs comprised another 25%. The molecular clock analysis estimated that HIV-1B was present in Bulgaria since the early 1980s, with a lower limit estimate for the first introduction in 1973 that is within the same time frame as the introduction of HIV-1 subtype B in North America. 27 In contrast to subtype B, the introduction of HIV-1 subtype A1 appeared to be more recent (early 1990s). Both molecular clock and viral gene flow analyses indicated a dynamic epidemic over the past 25 years, with multiple introductions from foreign countries and ongoing gene outflow from Bulgaria to West and East Europe. Although fewer than 600 cumulative AIDS cases have been identified in Bulgaria to date, our data suggest that the actual prevalence of HIV-1 in Bulgaria may be underestimated. The dynamic patterns of the HIV-1 epidemic within the country point out an enormous potential for spreading within the next few years, both within the country and to other neighboring countries in Europe. In this regard, three factors seem to be highly relevant: (1) the central geographic location of Bulgaria at the crossing between Western Europe, Eastern Europe, and the Middle East, (2) the high genetic heterogeneity of the HIV-1 epidemic in the Balkan countries revealed by the molecular characterization of our samples, and (3) the ongoing sociopolitical changes that have been affecting Bulgaria, as well as other countries in Eastern and Central Europe, since the end of the cold war.
The relatively few studies on the HIV-1 epidemic in other Eastern European countries have shown that non-B subtypes are quite common, with HIV-1A being highly prevalent in Russia, Belarus, and Ukraine, 28 –30 as well as the former Yugoslavia and Albania. 6,7 It has been demonstrated that shortly after the beginning of the subtype A epidemic in the FSU the virus was introduced into Ukraine and then spread, as shown in our analysis by the clustering of Bulgarian strains within the Eastern European clade, to other countries in Eastern Europe, 31 including Bulgaria. Surprisingly, only 1% of the subtype A1 Bulgarian strains actually clustered within the Eastern European clade. In fact, phylogenetic analysis indicated that at least three independent introductions of the HIV-1A1 subtype have occurred in Bulgaria, and that the majority of the HIV-1A Bulgarian strains in our sample were more closely related to West European subtype A sequences. Phylogeographic (migration) analysis showed that the largest HIV-1A1 outflow occurred from the African continent, a finding consistent with the African origin and the high seroprevalence of subtype A1 in Africa. 1 In addition, a significant HIV-1A1 gene flow from West Europe as well as other East European countries to Bulgaria was observed. The analysis revealed a complex web of introductions/transmissions of the virus between Bulgaria and other European countries, with at least four separate introductions of HIV-1B supported by both phylogeny and molecular clock techniques. The findings are consistent with the central geographic location of Bulgaria and point out that continuous surveillance of the epidemic in this Balkan country may be of great importance for the monitoring and prediction of future HIV-1 epidemiological trends in the rest of Europe.
A recent study explored the history of the HIV epidemic in the United Kingdom within defined risk groups. 32 The study revealed the complex epidemiological structure of different transmission clusters characterized by an initial period of exponential growth, followed by a slower spread as a consequence of behavior change within the risk group and the introduction of HAART. Most of the Bulgarian clades included viral strains from individuals infected exclusively through sexual contact. However, the presence of distinct and well-supported monophyletic clades within both HIV-1A1 and HIV-1B phylogeny suggests that after each initial introduction of the virus in a specific population, separate transmission clusters have been evolving along independent phylogenetic lineages. Better characterization and continuous monitoring of such groups are going to be crucial to understand in detail the epidemiology of HIV-1 in Bulgaria and to assess the efficacy of prevention and therapy in controlling the epidemic.
Some possible limits and biases of the present study should be mentioned. First, we cannot exclude the possibility that the study population is not truly representative of all HIV cases in Bulgaria. In particular, most persons included in our analysis had treatment failure; thus it is possible that persons in an advanced stage of disease (i.e., AIDS cases) were overrepresented. The possible consequences could be (1) underestimation of the increase in the circulation of different clades in recent infections or (2) overrepresentation of transmission clusters due to the elevated virulence of the circulating virus. Underestimation might occur only if changes in the circulation of different clades took place over recent years, which seems unlikely in the light of phylogeny and molecular clock analysis that showed a consistent evolution of independent transmission clusters after the initial HIV-1 introduction between 1980 and 1990. Overestimation is also unlikely to have affected our findings since there is no definitive evidence that some clades within a specific subtype (i.e., A vs. B) are more virulent than others, and no clear-cut association between a specific subtype and virulence has been reported, although increased virulence of subtype D over A has been noted. 33 Another possible limit of our study is use of subgenomic, rather than full genome, sequences, which could underestimate recombinant strains or overestimate subtype representation in the population. Even so, given the high statistical association of Bulgarian strains with specific Eastern and Western European sequences within both subtype A1 and B data sets, neither the conclusion of multiple HIV-1 introductions in Bulgaria nor the migration analysis was likely to be affected. In conclusion, we found a wide genetic diversity of HIV-1 in Bulgaria. Both subtype A1 and B appeared to have been introduced multiple times within the past two decades. Our data also suggest the extreme dynamic nature of the Bulgarian epidemic characterized by considerable viral inflow and outflow to/from other European countries, and emphasize the need for improved surveillance and control of HIV-1 infection in the Balkans.
Footnotes
Acknowledgments
The research was supported by PHS R01 awards HD032259, AI047723, and AI065265; General Clinical Research Center of the University of Florida MO1-RR00082; University of Florida Children's Miracle Network; Center for Research in Pediatric Immune Deficiency; Laura McClamma Research Fellowship; Pediatric Clinical Research Center of All Children's Hospital, University of South Florida, and the Maternal and Child Health Bureau, R60 MC 00003-01, Department of Health and Human Services, Health Resources and Services Administration; and Robert A. Good Chair for Immunology (J.W.S.) and Stephany W. Holloway University Chair for AIDS Research (M.M.G).