
Abstract
Ribonucleases are evoking medical interest because of their intrinsic cytotoxic activity. Most notably, ranpirnase, which is an amphibian ribonuclease, is in advanced clinical trials as a chemotherapeutic agent for the treatment of cancer. Here, we describe a strategy to create a novel antiviral agent based on bovine pancreatic ribonuclease (RNase A), a mammalian homologue of ranpirnase. Specifically, we have linked the N- and C-termini of RNase A with an amino acid sequence that is recognized and cleaved by human immunodeficiency virus (HIV) protease. This linkage obstructs the active site, forming an HIV-specific RNase A zymogen. Cleavage by HIV-1 protease increases ribonucleolytic activity by 50-fold. By relying on the proper function of HIV-1 protease, rather than its inhibition, our approach will not engender known mechanisms of resistance. Thus, we report an initial step toward a new class of agents for the treatment of HIV/AIDS.
Introduction
S
This slowing of progress against HIV/AIDS, combined with several major problems with HIV/AIDS therapy, continues to motivate the design of new drugs. 8,9 Side effects, especially disturbances in lipid metabolism, are an increasing problem as patients are living longer and developing cardiac disease. 2,10 Rapid viral replication and the high error rate of reverse transcriptase have led to high rates of resistance to HAART, 5 with about 10% of newly acquired infections in the United States and Europe being resistant to at least one of the three major drug classes. 11 The rapid development of resistance necessitates strict patient compliance with therapy. Finally, AIDS remains a chronic disease due to the existence of latent HIV infection in memory T cells and other cells that are not pharmacologically accessible. 12,13
One strategy for slowing viral progression and eliminating latent viral infection is to kill those cells that are infected with HIV. Although cytotoxic cancer drugs could be used, there is a risk of serious side effects that would compound the side effects of HAART. 14 To circumvent this problem, toxins that are specific for HIV-infected cells have been designed. One approach has been to use toxins that will be activated by the HIV-1 protease (HIV PR), a virally encoded protein that cleaves the viral polyprotein late in the life cycle of HIV. 15 In one case, an HIV PR-activated variant of caspase-3 was constructed and its ability to kill HIV-infected cells was demonstrated. 16 In another case, diphtheria toxin was modified such that a degradation signal was removed by cleavage with HIV PR. 17 These “pro-drug” strategies have the potential to reduce the problems with both side effects and drug resistance.
Bovine pancreatic ribonuclease (RNase A) has been studied intensely by biochemists for decades. 18,19 Recently, interest in RNase A has resurged with the discovery of the facinating biological properties of ranpirnase, angiogenin, and bovine seminal ribonuclease, which are homologues of RNase A. 20 –25 For example, ranpirnase 26 is toxic to cancer cells in vitro and in vivo in a manner that is dependent on its catalytic activity, 27 and is currently in Phase IIIb clinical trials for the treatment of malignant mesothelioma. 28 In addition, RNase A and other ribonucleases are known to inhibit HIV replication. 29,30
Here, we describe the creation of an HIV-specific zymogen from RNase A. Inspired by the design of zymogens specific for malaria and hepatitis C, 31,32 we sought to exploit the activity of HIV PR to activate a cytotoxin. The N- and C-termini of RNase A were joined by circular permutation, thereby occluding the active site with an amino acid sequence containing a cleavage site for HIV PR (Fig. 1). In the presence of HIV PR, this sequence is cleaved and the catalytic activity is unmasked. It is our hope that because a chemotherapeutic strategy based on a ribonuclease zymogen would rely on catalysis by HIV PR rather than merely an affinity for that enzyme, the development of resistance would be unlikely.

Design of an HIV-specific ribonuclease A zymogen. Circular permutation was used to block access to the active site until activation by HIV PR.
Materials and Methods
Materials
Escherichia coli strain BL21(DE3) was from Novagen (Madison, WI). K-562 cells, 33 which are human erythroleukemia cells, were obtained from American Type Culture Collection (Manassas, VA). HIV PR expression vector pET-HIVPR 34 was a kind gift from J. Tang (Oklahoma Medical Research Foundation).
Enzymes were obtained from Promega (Madison, WI). Protein purification columns and resin were from Amersham Biosciences (Piscataway, NJ). Synthetic oligonucleotides, including the ribonuclease substrate 6-FAM-dArU(dA)2-6-TAMRA, 35 were from Integrated DNA Technologies (Coralville, IA). Poly(cytidylic acid) [poly(C)] was from Sigma–Aldrich (St. Louis, MO) and was precipitated with ethanol before use to remove short RNA fragments. [methyl-3H]Thymidine was from Perkin–Elmer (Boston, MA). MES buffer (Sigma-Aldrich, St. Louis, MO) was purified by an-ion-exchange chromatography to remove any contaminating oligo(vinylsulfonic acid) (OVS). 36 All other chemicals were of commercial grade or better, and were used without further purifications.
Phosphate-buffered saline (PBS) contained (in 1 liter) NaCl (8.0 g), KCl (2.0 g), Na2HPO4 ∙ 7H2O (1.15 g), and KH2PO4 (2.0 g), and had a pH of 7.4.
Zymogen expression and purification
Plasmids used to direct the production of HIV RNase A zymogens were derived from the plasmid pET22b(+)/19N.
31
The region of the plasmid that encoded the linker was replaced with DNA encoding the sequence GSTATIMMQRGNAG (Zymogen
Values of T m were determined in PBS by UV spectroscopy.
Value of K d (± SE) was determined for the complex with hRI at (23 ± 2)°C.
Values of IC50 are for the incorporation of [methyl-3H]thymidine into the DNA of K-562 cells.
Values of m/z were determined by MALDI-TOF mass spectrometry.
From Rutkoski et al. 42
From Lee et al. 53
Protease expression and purification
The substitutions Q7K/L33I/L63I
37
and C67A/C95A
38
were made with the Quikchange mutagenesis kit to improve protease stability. The resulting plasmid, pET-HIVPRV, was transformed into E. coli BL21(DE3) cells for expression. Transforming, plating, and subculturing were done in Luria–Bertani medium containing glucose (1% w/v) to reduce leaky expression of HIV PR, which is toxic to E. coli.
39
Protease expression was induced in Terrific Broth containing isopropyl-β-
Zymogen cleavage
Zymogens were activated by mixing them with 0.04–0.2 M equivalents of HIV PR in reaction buffer, which was 100 mM sodium acetate buffer, pH 5.0, containing NaCl (100 mM), EDTA (1 mM), glycerol (5% v/v), and PEG-8000 (0.1% w/v), and incubating the resulting solution at 37°C for 1 h. Activation was stopped by dilution into gel-loading buffer to a final composition of 50 mM Tris–HCl, pH 6.8, containing DTT (100 mM), SDS (0.2% w/v), glycerol (10% v/v), and bromophenol blue (0.75 mM), or by dilution (by 100-fold) into 0.10 M MES–NaOH, pH 6.0, containing NaCl (0.10 M). Following dilution, reaction mixtures were placed on ice. SDS-PAGE in the presence of DTT was used to assess zymogen cleavage (Fig. 2).
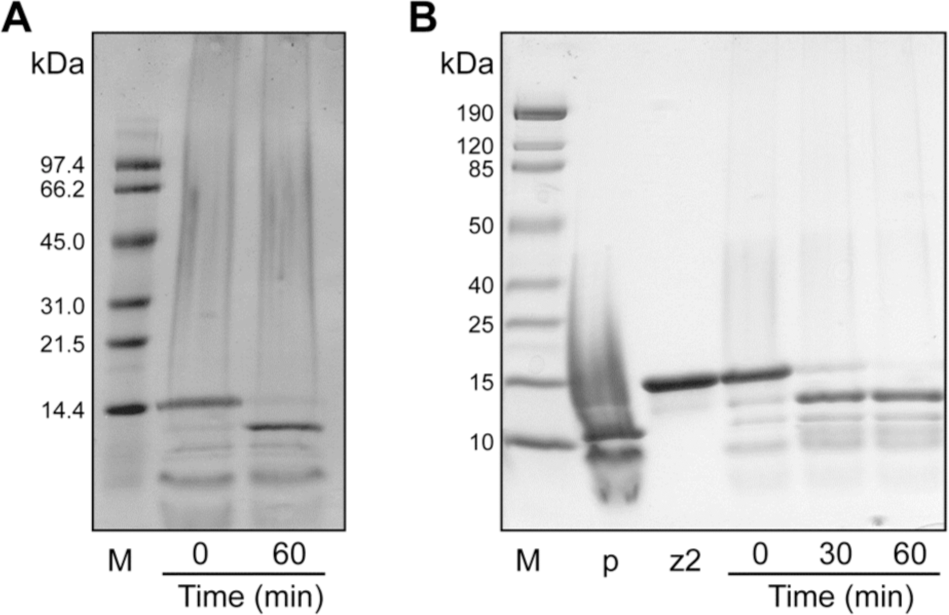
Activation of zymogens by HIV PR. Activation at 37°C was monitored by SDS–PAGE in the presence of DTT. (
Ribonucleolytic activity
The ability of zymogens to catalyze the cleavage of the fluorogenic substrate 6-FAM-dArU(dA)2-6-TAMRA, which exhibits a 180-fold increase in fluorescence (excitation at 493 nm; emission at 515 nm) upon cleavage,
35
was assessed. Assays were carried out at (23 ± 2)°C in 2.0 ml of 0.10 M MES–NaOH (OVS-free), pH 6.0, containing NaCl (0.10 M), substrate (20 nM), and zymogen (5 pM–1 nM). Values of k
cat/K
M were obtained with Eq. (1):
where ∆I/∆t represents the initial reaction velocity, I max is the fluorescence following complete cleavage of the substrate by excess RNase A, and I 0 is the fluorescence prior to addition of ribonuclease.
In addition, the ability of zymogens to cleave poly(C) (ɛ 268nm = 6200 M−1 cm−1 per nucleotide) was monitored by the change in UV absorption, which increases upon cleavage (∆ɛ 250 nm = 2380 M−1 cm−1). Assays were carried out at (23 ± 2)°C in 0.10 M MES–NaOH (OVS free), pH 6.0, containing NaCl (0.10 M), poly(C) (10 μM–1.5 mM), and zymogen (20 nM–1 μM). Initial velocity data were used to calculate values of k cat, K M, and k cat/K M with the program Prism 4 for Macintosh (GraphPad Software, San Diego, CA).
Conformational stability
The value of T m, which is the temperature at the midpoint of the thermal transition between the folded and unfolded states, of each zymogen was determined by monitoring its UV absorption at 287 nm. Zymogen solutions (∼25 μM in PBS) were heated incrementally (0.15°C/min from 25 to 75°C). Data were collected and analyzed with the program THERMAL from Varian Analytical Instruments (Walnut Creek, CA).
Inhibition by the ribonuclease inhibitor protein
Binding of unactivated zymogen
Cytotoxic activity
K-562 cells were grown in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml). The effect of zymogens on K-562 proliferation was measured by the incorporation of [methyl-3H]thymidine as described previously. 42
Results
Design of HIV-specific zymogen
The p2/NC recognition sequence of HIV PR (TATIM/MQRGN) was chosen because it is cleaved initially and efficiently in vitro and in vivo.
43,44
It is desirable for a zymogen to contain a linker that is long enough to allow for the flexibility necessary to be cleaved by the protease, but also short enough to prevent indiscriminate ribonucleolytic activity prior to activation by the specific protease. We designed two HIV-specific RNase zymogens, one with a 14-residue linker (zymogen
Circular permutation was done to produce new N- and C-termini at residues 89 and 88 of RNase A (Fig. 1). These two residues were linked by a new disulfide bond. 31 In addition, residues 4 and 118 (RNase A numbering) were replaced with cysteine residues and linked by a disulfide bond in order to improve stability. 45
Zymogen activation
Zymogens
The ability of zymogens to cleave two RNA substrates was measured (Tables 2 and 3). The shorter substrate was a fluorogenic tetranucleotide that increases in fluorescence upon cleavage (Table 2).
35
The catalytic efficiency (k
cat/K
M) of zymogen
Values of K cat /K M (± SE) were determined for catalysis of 6-FAM-dArU(dA)2-6-TAMRA cleavage at 25°C in 0.10 M MES–NaOH buffer (OVS free), pH 6.0, containing 0.10 M NaCl.
N/A, not applicable.
From Rutkoski et al. 42
Values of k cat, K M, and k cat/K m (± SE) were determined for catalysis of poly(C) cleavage at 25°C in 0.10 M MES–NaOH buffer (OVS free), pH 6.0, containing 0.10 M NaCl. Initial velocity data were used to calculate values of k cat, K M, and k cat/K M with the program Prism 4 (GraphPad Software, San Diego, CA).
From Johnson et al. 32
N/A, not applicable.
This sensitive assay for ribonucleolytic activity might not be an accurate reflection of the ability of ribonucleases to cleave therapeutically relevant substrates.
47
Hence, we measured the ribonucleolytic activity of zymogen
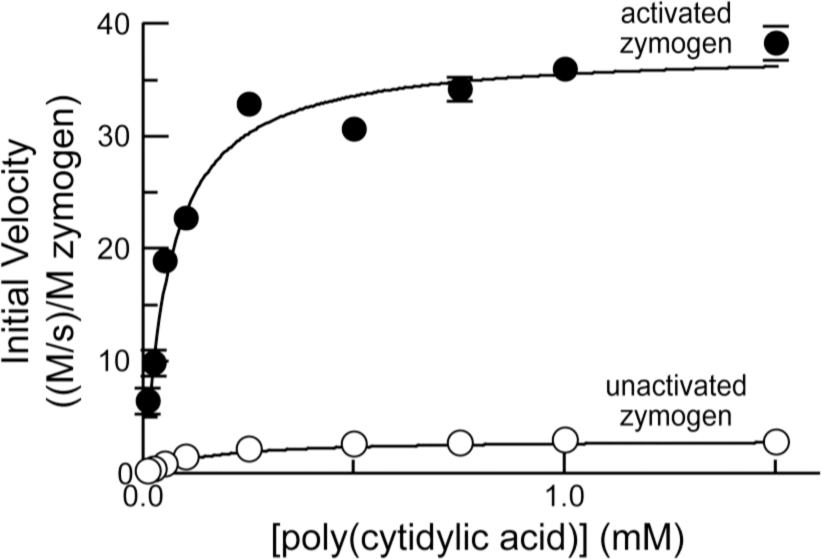
Enzyme kinetics of unactivated zymogen
Conformational stability of zymogens
The ability of a cytotoxic ribonuclease to retain its conformation at physiological temperatures is an important factor in determining its potential as a chemotherapeutic agent.
45
Zymogens
Binding of ribonuclease inhibitor protein
The ability of a ribonuclease to evade binding by the cytoplasmic ribonuclease inhibitor protein (RI), which binds RNase A with femtomolar affinity, is essential for its cytotoxicity. 42,48,49 The active site of the ribonuclease comprises a large portion of the buried surface area in the ribonuclease–inhibitor complex. 50,51 We would expect that the unactivated RNase A zymogen would have diminished affinity for RI, and indeed the K d value for the ribonuclease–RI complex with the unactivated zymogen is >106-fold greater than that for wild type RNase A (Table 1).
Cytotoxicity of unactivated zymogen
A cytotoxic therapeutic agent must have little or no toxicity to healthy cells—those that are not infected with HIV. To assess this attribute, unactivated zymogens were assayed for their ability to inhibit the proliferation of K-562 cells, which are human cells that are not infected with HIV. Neither zymogen
Discussion
We have created two HIV-specific RNase A zymogens. Their activation by HIV PR is 95% complete following a 1-h incubation with a substoichiometeric amount of HIV PR (Fig. 2). One factor that determines the efficiency of this cleavage reaction is the ability of the protease to bind to the zymogen. HIV PR and zymogens
The two zymogens differ as catalysts of RNA cleavage. The ribonucleolytic activity of zymogens
The use of a longer RNA substrate revealed additional information. Using poly(C) as a substrate, unactivated zymogen
The development of a therapy that kills HIV-infected cells is highly attractive. Such a therapy could eradicate completely the viral reservoir in patients, a challenge that remains in the era of HAART. 13 Drugs that are used to kill cancer cells could be used for this purpose but would result in the side effects typical of cancer chemotherapy, including bone marrow suppression. Any potential for decreasing white blood cells in a patient with a weakened immune system should be avoided. As an alternative, our strategy seeks the selective destruction of HIV-infected cells.
Ribonuclease zymogens have other desirable attributes. Ribonucleases that evade the endogenous ribonuclease inhibitor protein are known to be potent cytotoxins. 49 In contrast, we have shown that ribonuclease zymogens have no observable toxicity toward a human cell line that does not contain HIV PR (Table 1). This absence of cytotoxicity suggests that a drug based on a ribonuclease zymogen could have a high therapeutic index. Another attractive feature is the potential difficulty in the development of viral resistance to a ribonuclease zymogen. Rapid viral replication and the high error rate of reverse transcriptase have led to the rapid development of resistance to reverse transcriptase inhibitors and protease inhibitors, small-molecule drugs that rely on binding to these enzymes. It is reasonable to anticipate that resistance to a drug that requires the catalytic activity of HIV PR toward a native substrate would be significantly more difficult for a virus to develop. Moreover, resistance derived from a protease variant with altered specificity could be countered by replacing the linker sequence with one that is recognized by the protease variant. Finally, we note that a chemotherapeutic agent based on a ribonuclease zymogen would not be used concurrently with protease inhibitor therapy, which would likely diminish zymogen activation and hence efficacy.
In conclusion, we have described the creation of an HIV-specific RNase A zymogen that increases in catalytic efficiency by approximately 50-fold upon activation with HIV PR, is stable at physiological temperature, and is not toxic to uninfected cells. These data establish a proof of principle for a new class of agents for the treatment of HIV/AIDS. We anticipate testing our strategy with human cells containing HIV PR.
Footnotes
Acknowledgments
We are grateful to J.F. May and S.M. Fuchs for assistance with cloning, L.D. Lavis for the diethylfluorescein derivatives, used in the microplate assay, and M.N. Levine, S.M. Fuchs, K.A. Dickson, and R.J. Johnson for contributive discussions. This work was supported by Grants 51670 (Bill & Melinda Gates Foundation) and CA073808 (NIH). R.F.T. was supported by a Wisconsin Distinguished Rath Graduate Fellowship. The University of Wisconsin–Madison Biophysics Instrumentation Facility was established with grants BIR-9512577 (NSF) and RR13790 (NIH). The Keck Center for Chemical Genomics was established with a grant from the W.M. Keck Foundation.
Disclosure Statement
No competing financial interests exist.