
Abstract
The molecular epidemiology of HIV-1 infection was previously studied in Cyprus but the degree of HIV-1 diversity has remained indefinable. The main objective of the present study is to examine HIV-1 strains isolated from 77 HIV-1-infected individuals representing 38% of the known infected population in Cyprus in the period 1986 to 2006. DNA of the near full-length genome encoding gag, pol, vif, vpr, vpu, tat, rev, env, and 5′-end of nef was amplified by nested PCR/RT-PCR from all HIV-1 seropositives and sequenced using a newly designed assay. Detailed phylogenetic and bootscanning analyses were performed to determine phylogenetic associations and subtype assignments. Phylogenetic analyses of the obtained viral sequences indicated that subtype B was the dominant subtype (61%), followed by subtype A (23.3%), subtype C (5.2%), CRF02_AG (3.9%), and subtype D, CRF01_AE, and CRF04_cpx (1.3% each). Two HIV-1 isolates (2.6%), originating from the Democratic Republic of Congo (DRC), were not classified in any pure (sub)subtype or circulating recombinant form (CRF). Complete phylogenetic and bootscanning analyses revealed that one of these isolates had a new, unique recombinant pattern, comprising segments of subtypes D and G, and is distinct from any other CRFs or URFs reported so far. Detailed analyses of the sequence of the second isolate, which could not be classified, reveal that it is close to subtype K reference sequences but clusters near the root of the clade. At least two epidemiologically unrelated HIV-1 seropositives with an HIV-1 variant similar to this isolate are required to designate this variant as a novel HIV-1 subtype or subsubtype of subtype K. Analogous to results of the earlier epidemiological studies, these data exhibit the extensive heterogeneity of HIV-1 infection in Cyprus, which is being fueled by a continuous entry of new strains from other countries, creating an evolving and polyphyletic infection.
Introduction
H
The first AIDS patient in Cyprus was reported in 1986 and in 1994 the first molecular epidemiological study of HIV-1 infection in Cyprus was carried out. 19 The study was based on phylogenetic analysis of viral sequences encoding the C2 to V3 env gp120 region from 24 patients infected from 1987 to 1994. Subtype B was found to be the dominant strain accounting for 63% of the infections, followed by subtypes C and I (later determined as CRF04_cpx 20 ) 4% each, and two distinct clusters in which one was related to subtype A (21%) and the other to subtype F (8%). 19 This subtype heterogeneity was confirmed in a subsequent study, 21 where HIV-1 strains from 37 newly diagnosed untreated HIV-1 patients in the period 2003 to 2006 were characterized based on phylogenetic analyses of obtained viral sequences encoding the gag region, the pol (protease and reverse transcription, RT), and the env (gp160). Subtype A was identified as the most prevalent subtype accounting for 38% of the newly diagnosed infections, followed by subtype B (35%), subtype C (13%), CRF02_AG (8%), and subtypes D and CRF01_AE (3% each). The diversity of HIV-1 subtypes in Cyprus is extensive and led us to assume that HIV-1 isolates from Cyprus might be uncommonly heterogeneous, including new variants or recombinant forms. As part of a growing effort to monitor and characterize the molecular epidemiology of HIV in Cyprus, in this study we determined the genetic diversity among HIV-1 strains isolated from 77 HIV-1 seropositives diagnosed from 1986 to 2006 by means of a near full-length genome sequence analysis.
Materials and Methods
Study subjects
For the period 2003 to 2006 blood samples were obtained from 77 consenting HIV-1-infected individuals from the Cyprus Reference AIDS Clinic of Larnaca National Hospital. All HIV-1 study subjects were diagnosed from 1986 to 2006 and represent 38% of the living known infected population diagnosed from 1986 to 2006. The majority of study subjects were Greek-Cypriots, although a number reported traveling or living abroad in the past. All blood samples were processed at the Laboratory of Biotechnology and Molecular Virology of the University of Cyprus within the same day of sampling.
PCR amplification of near-full genome and sequencing
Patient's blood (16 ml) was collected in CPT tubes (Becton Dickinson, Annapolis, MD); peripheral blood mononuclear cells (PBMCs) and plasma were isolated using the CPT vacutainer procedure. HIV-1 RNA was extracted from 200 μl of plasma and genomic DNA was extracted from about 107 uncultured PBMCs using the QIAamp UltraSens Virus Kit and QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA), respectively. HIV-1 sequences encoding approximately 1722 bp of the gag region, 1461 bp of the pol (protease and RT) region, 3118 bp of the pol (RNase H, integrase IN) region and vif, vpr, and vpu genes, and 2927 bp of the env (gp160) and 5′-end of the nef region were amplified from each sample by nested polymerase chain reaction (PCR) applying PBMC-associated HIV-1 DNA (Fig. 1A, B, C). For any samples in which the PCR product of genomic DNA was problematic, reverse transcription nested PCR (RT-PCR) using plasma HIV-1 RNA was performed instead. Primers used in the PCR and/or RT-PCR reactions are described in Table 1 and their positions correspond to the HXB2 strain (accession number K03455). Reagents and thermocycling profiles for the amplification of each region have been described elsewhere. 21,22 Amplified products from the second-round PCR were purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA). Their lengths were analyzed by 1% agarose gel electrophoresis and the concentrations were quantified by UV absorbance spectrophotometry using the Nanodrop ND-1000 Spectrophotometer (Nanodrop Technologies, Wilmington, DE). In each sample, the DNA sequences encoding the gag, pol (protease and RT), and pol (RNase H and IN) region and vif, vpr, and vpu genes and the env (gp160) region and 5′-end of nef were determined by direct sequencing in separate reactions, using second-round-amplified PCR product as the template and sequencing primers (Table 1, Fig. 1D). Reagents and thermocycling profiles used in each amplification reaction of sequencing are described elsewhere. 21,22 Amplicons were sequenced with the ABI 3130 genetic analyzer (Applied Biosystems, Foster City, CA). Samples exhibiting partial or extensive viral diversity by direct sequencing were subsequently cloned, using the TOPO TA cloning kit for sequencing (Invitrogen Corp., San Diego, CA), and sequenced as described. The sequence data were analyzed and edited using the Sequencing analysis 5.2 program (Applied Biosystems, Foster City, CA) and the Primer Premier 5 application (Primer Biosoft International, Palo Alto, CA). The sequence fragments of each of the four amplified regions were assembled manually, with overlapping segments removed to acquire the near full-length sequences for each sample.
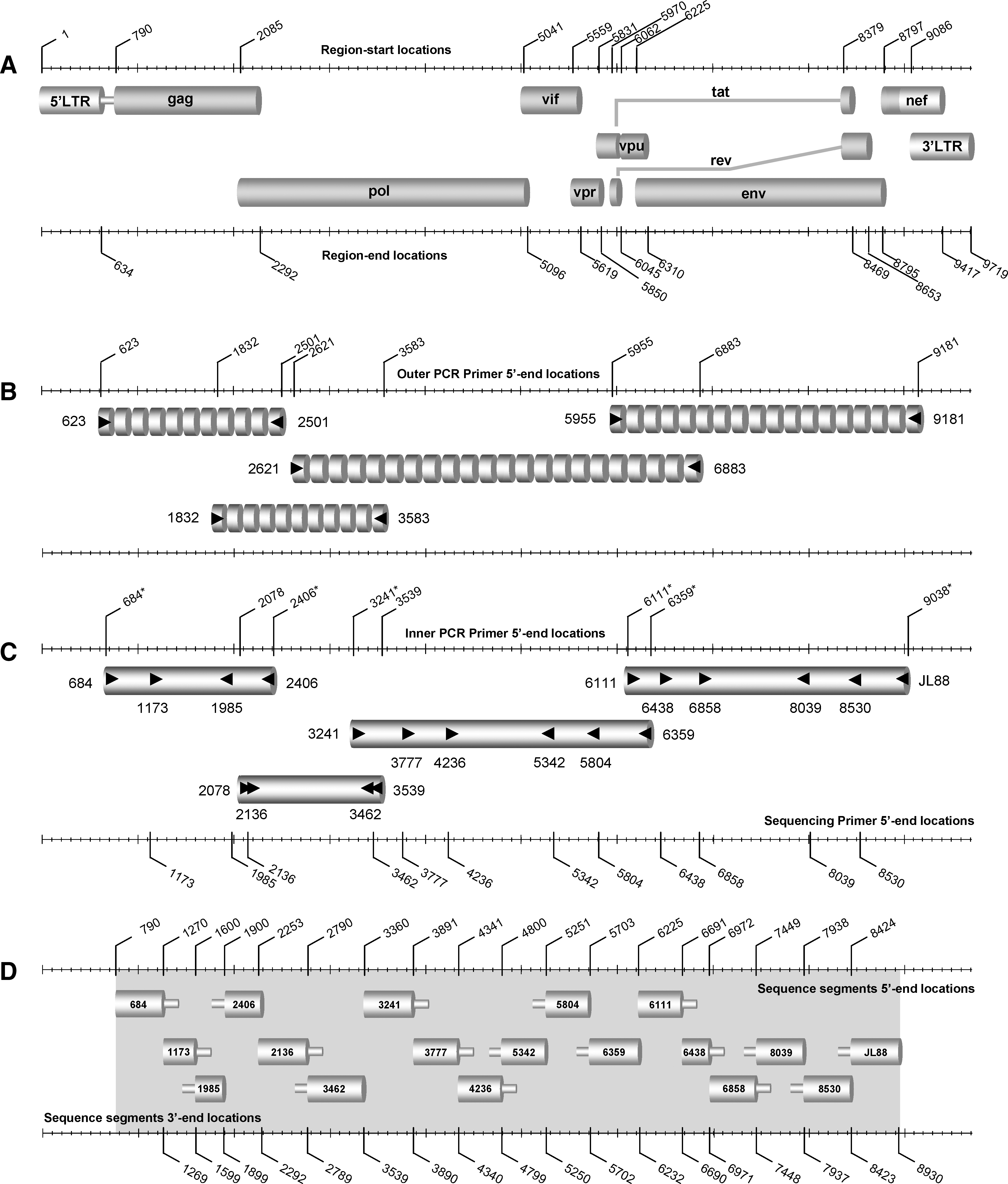
Schematic representation summarizes the amplification of the new full-length genome region within HIV-1 M group subtypes used in an assay described in this study. (
Reverse transcriptase, primary and secondary PCR primers, and sequencing primer names as these appear in the text; the orientation of the PCR primer is indicated in parenthesis: F, forward; R, reverse.
Y, indicates equal molar mixture of C and T; R, A, and G; M, A, and C; W, A, and T; V, A, C, and G.
Primer positions correspond to subtype B HIV-1 HXB2 strain (accession number K03455).
Used also as sequencing primers as described in the text.
Phylogenetic analysis
Bionumerics 5.0 program 23 was used for multiple DNA alignment. The Molecular Evolution Genetic Analysis (MEGA) software was used for distance calculation and phylogenetic tree construction. 24 Study subjects' DNA sequences encoding near full-length viral regions were aligned against corresponding near full-length genome sequences of genetically characterized HIV-1 strains obtained from the Los Alamos database. 5 Pairwise distance matrices were calculated using the Kimura two-parameter distance estimation approach with a transition/transversion ratio of 2.0 and phylogenetic trees were constructed using neighbor joining (NJ). The consistency of the phylogenetic clustering was tested using bootstrap analysis with 1000 replicates. For one isolate the phylogenetic analysis was confirmed by using the maximum-likelihood (ML) model with the Jukes-Cantor evolutionary model. Bootstrap values above 70 were considered adequate for subtype assignment. The subtype assignment was confirmed using the REGA algorithm. 25 GenBank accession numbers for control reference sequences in phylogenetic analyses are as follows: A1-PS1044, DQ676872; A1-92UG037, AB253429; A1-94Q2317, AF004885; A1-92RW008, AB253421; A2-97CDKTB48, AF286238; A2-94CY01741, AF286237; A2-97CDKS10, AF286241; B-83HXB2.LAI.IIIB.BRU, K03455; B-981058, AY331295; B-90BK132, AY173951; B-0067100T36, AY423387; B-98153841, DQ853463; C-92BR025d, U52953; C-86ETH2220, U46016; C-04SK164B1, AY772699; C-95IN21068, AF067155; D-01A280, AY253311; D-94UG114, U88824; D-83ELI, K03454; D-01CM4412HAL, AY371157; F1-93VI850, AF077336; F1-93FIN9363, AF075703; F1-9393BR020-1, AF005494; F1-96MP411, AJ249238; F2-95MP255, AJ249236; F2-95MP257, AJ249237; F2-02CM0016BBY, AY371158; F2-97CM53657, AF377956; G-93HH8793121, AF061641; G-96DRCBL, AF084936; G-92NG083, U88826; G-PT2695, AY612637; H-93VI997, AF190128; H-90056, AF005496; H-93VI991, AF190127; J-94SE7022, AF082395; J-93SE7887, AF082394; J-97DCKTB147, EF614151; K-97EQTB11C, AJ249235; K-96MP535, AJ249239; 01AE-90CM240, U54771; 01AE-93TH051, AB220944; 02AG-IBNG, L39106; 02AG-99pBD615, AY271690; 04cpx-97PVMY, AF049337; and 04cpx-9197PVCH, AF049292.
Analysis of the intersubtype mosaicism
To explore putative recombination patterns in the sequences we performed a bootscanning analysis using Simplot, version 3.5.1, 26 which is a sliding window approach that allows identification and evaluation of the putative intersubtype recombination breakpoints of the query sequence by the graphic detection of a change in the phylogenetic signal. For the query sequence we run similarity and bootscanning analyses against a pure subtype reference set. Bootscanning was performed with a sliding window of 400 nucleotides overlapped by 40 nucleotides to define the recombinant structure. Subregion confirmatory NJ tree analyses were carried out using MEGA software 24 to confirm the subtype origin within each gene fragment. Bootstrap analysis (1000 replicates) was used to estimate the reliability of the constructed trees and a bootstrap value of 70% was considered to be definitive. To look for any potential relationships between possible recombinant sequences and previously characterized HIV-1 sequences, a BLAST search was performed using the default settings in the HIV BLAST tool available at the Los Alamos HIV Sequence Database. 27,28
Results
Clinical and epidemiological features of the study subjects
The study group consisted of 77 HIV-1 seropositives and each subject is identified by a laboratory registration number ascending in chronological order of blood drawn. The study subjects represent 38% of the HIV-1-infected individuals diagnosed from 1986 to 2006. Blood samples from all study subjects were withdrawn in the period from 2003 to 2006. The general demographic, epidemiological, and clinical features of all study subjects are summarized in Table 2. Among the 77 study subjects, 56 (72.7%) were male and 21 (27.3%) were female. Their median age was 38 years and interquartile range (IQR) 32 to 47. The median CD4+ lymphocyte count was 448 cells/mm3 (IQR, 232 to 651) and the median plasma HIV load was 3.59 log HIV-1 RNA copies/ml (IQR, 1.7 to 4.6). Fifty-six HIV-1 seropositives are Cypriots (72.7%); seven are from sub-Saharan Africa (9.1%), seven, from eastern Europe (9.1%), six from western Europe (7.8%); and one (1.3%) from the United States. Thirty-seven study subjects (48.0%) were infected by homosexual/bisexual contact, 39 (50.6%) by heterosexual contact, and one (1.3%) by intravenous drug use. Thirty-one subjects (40.2%) were infected in Cyprus, 17 (22.1%) in western Europe, 4 (5.2%), in eastern Europe, 11 (14.3%), in countries of sub-Saharan Africa, 2 (2.6%) in the United States, and 1 (1.3%) in southeast Asia. For 11 study subjects (14.3%) the country in which the infection was most likely contracted is unknown. Among study subjects there are four heterosexual couples, one infected in the UK, one in Georgia, the other in Cyprus, and the last with an unknown place of infection.
Indicates the laboratory code for each study subject.
F, female; M, male.
Indicates the date (month/year; –, unknown month) of the first known positive HIV antibody test.
Country of birth of the study subjects.
MSM, men who have sex with men; HSX, heterosexual contact; OHPC, origin from a high prevalence country; IVDU, intravenous drug user.
Information provided by the study subjects. N/A, Not available; CMV, Cytomegavirus; HBV, Hepatitis B virus; HCV, Hepatitis C virus.
Sequence analysis of near full-length HIV-1 genome
Uncultured PBMCs and plasma from all subjects were HIV-1 positive by nested PCR and/or RT-PCR in gag, pol (protease and RT), pol (RNase H and IN), and vif, vpr, and vpu genes, env (gp160), and the 5′-end of nef regions. The positive PCR combined with the extensive genetic diversity of HIV-1 strains, as described in the phylogenetic analysis (Fig. 2), demonstrates that the designed PCR primers (Table 1) are suitable or diverse M-group strains. All HIV-1 internal PCR products were further analyzed by nucleotide sequencing analysis using sequencing primers for each region (Table 1, Fig. 1D). The near full-length genome of the HIV-1 strains was assembled from the overlapping sequenced fragments. The complete near full-length DNA sequences encoding the expected nine open reading frames for gag, pol, vif, vpr, vpu, tat, rev, env, and the 5′-end of nef (Fig. 1A) were successfully derived from all 77 study subjects.

NJ phylogenetic tree for the nucleotide sequence of the near full-length genome of HIV-1 strains obtained from 77 HIV-1-seropositive patients in Cyprus, based on the Kimura two-parameter distance estimation method. The tree includes representative reference sequences of HIV-1 subtypes (A–K) and CRF01_AE, CRF02_AG, and CRF04_cpx (shown in bold). The numbers indicated at several nodes are consensus bootstrap values out of 1000 replications (only bootstrap values greater than 50% are denoted). The sequences determined in the study are shown in black boxes with a prefix CY for Cyprus and the numbers following denoting the laboratory code. The unclassified sequences are shown in boxes. Epidemiologically linked patients are indicated by brackets. The divergence between any two sequences is obtained by summing the horizontal branch length, using the scale at the lower left, which represents 2% genetic distance (0.02 substitutions per site). The brackets on the right side of the tree indicate the determined subtypes as described in Results; U, for unclassified.
Phylogenetic analysis
The molecular epidemiological relationships among DNA sequences encoding near full-length genomes (790 to 8930 nt, Fig. 1A, D) were analyzed by nucleotide phylogenetic analysis. One phylogenetic tree was constructed for the 77 study subjects on the basis of 77 derived near full-length DNA sequences (Fig. 2). In addition to the sequences from Cyprus, 46 previously sequenced HIV-1 isolates from diverse global locations, encompassing all nine known subtypes (A through K) and three CRFs (CRF01_AE, CRF02_AG, and CRF04_cpx), were also included in the analysis. According to the constructed NJ phylogenetic tree shown in Fig. 2, four distinct subtypes (A, B, C, and D) and three CRFs (CRF01_AE, CRF02_AG, and CRF04_cpx) were identified for the Cypriot sequences within the M group: subtype A, 18 sequences (23.3%); subtype B (61.0%), 47 sequences; subtype C, 4 sequences (5.2%); subtype D, 1 sequence (1.3%); CRF01_AE, 1 sequence (1.3%); CRF02_AG, 3 sequences (3.9%); and CRF04_cpx, 1 sequence (1.3%). It is important to note that the Cypriot sequences in subtypes A, B, C, and CRF02_AG have a relatively high average intrasubtype genetic diversity. The average (range) intrasubtype nucleotide divergence among the Cypriot gag sequences with subtype A is 11.0% (1.4–14.8%); within subtype B, 10.3% (4.4–19.4%); within subtype C, 10.0% (0.9–10.8%); and within CRF02_AG, 11.6% (8.5–13.5%). This variation is within the range of intrasubtype diversity and suggests that subtypes A, B, C, and CRF02_AG were transmitted to Cyprus during the study period (1986 to 2006) by multiple sources, which is consistent with the epidemiological data of the study subjects presented in Table 2.
The study group consisted of four heterosexual couples: a couple from Georgia (CY057 and CY058) infected in Georgia; a couple, the man from UK (CY112) and the woman from Sweden (CY111), with site of infection unknown; and two couples from Cyprus, one (CY088 and CY089) infected in Cyprus and the other (CY037 and CY038) in the UK. In the phylogenetic tree the sequences of three of the couples are very closely clustered: CY057–CY058 and CY111–CY112 in subtype A and CY088–CY089 in subtype B, with bootstrap values of 100%. The branch topologies in the phylogenetic tree (Fig. 2) reconfirm this relation. The average intrasubtype diversity between the gag sequences of CY111–CY112 is 1.5% and for the sequences CY057–CY058 is 0.7%; for the sequences CY088–CY089 it is 1.9%. The relatively low genetic diversity between these sequences in subtype A and subtype B, in comparison with the overall intrasubtype diversity for A (11%) and B (10.3%), respectively, reconfirms that the isolates were derived from epidemiologically linked individuals. The average intrasubtype diversity between the gag sequences of couple CY037 and CY038 (subtype B) is 11.0% and the branch lengths of the tree indicate that although they are epidemiologically linked they do not cluster very closely.
Two sequences (2.6%), isolated from CY063 and CY090 HIV-1 seropositives, do not cluster within pure subtypes or CRFs. As indicated in Fig. 2, the sequence of the CY063 isolate is branched close to subtype G reference sequences with a considerable bootstrap value of 100%, whereas the genetic distance of the CY090 strain sequence is closest to subtype K variants with a bootstrap value of 95%. The HIV-1 patient CY063 originating from the Democratic Republic of Congo (DRC) was infected before 1995 in the DRC by heterosexual contact. Running the CY063 sequence with the REGA algorithm, a recombinant pattern between subtypes G and D was indicated (data not shown). The HIV-1-infected patient CY090 originating from Cyprus was infected before 1993 in the DRC, by heterosexual contact. The sequences from these two isolates seem to be a unique case.
Evidence of mosaicism
For a more detailed comparison with other subtypes of HIV-1, the two unclassified sequences CY063 and CY090 were analyzed by similarity and bootscanning analysis. The similarity plot and the bootscan analysis showed that the genome of the CY063 sequence consisted of segments clustering alternatively with references of subtype G and D (Fig. 3). In the bootscan analysis a high bootstrap value (≥70%) was indicated in almost all regions of the genome, supporting the classification as a recombinant strain. Subtype assignments for each segment derived from the bootscan analyses were confirmed with NJ trees, in each case with a bootstrap value of ≥70% supporting the relation with subtype references (Fig. 4). The CY063 sequence contained 10 breakpoints (Fig. 3). The beginning of the genome, nucleotide (nt) 790–3000 (HXB2 numbering), was subtype G with a bootstrap value of 100%, shifting to subtype D with a low bootstrap value of 60% in the 3′-end of RT and 5′-end of RNase H of pol (3000–4100 nt). The third subregion in the 3′-end of RNase H and 5′-end of IN of pol (4100–4550 nt) clustered with subtype G with an 89% bootstrap value, followed by a small fragment in IN (250 nt) that was an area of poor resolution between subtypes D and G (Fig. 3). This area clustered with reference sequences of subtype D at a low bootstrap value of 11% in the NJ and was considered as unclassified. From the 3′-end of IN to the 3′-end of vpr (4800–5700 nt) the genome was of subtype G with the bootstrap value of 97%. The genome was shifted back to subtype D (5700–6300 nt) with an 81% bootstrap value and had the next breakpoint at the beginning of env (gp120). A following small fragment of 150 nt clustered with reference sequences of subtype G with a bootstrap value 86%. The eighth fragment (6450–7200 nt) clustered in the NJ tree with subtype D and a 93% bootstrap value, while the next subregion (7200–7600 nt) of env (gp120) was determined as unclassified as it was poorly resolved between D and B references, and it was clustered in the NJ tree with subtype B with a 65% bootstrap value. The tenth fragment (7600–8700 nt) coding part of exon 2 of rev and tat through env (gp41) belonged to subtype D with a bootstrap value of 95%. The last region (8700–8930 nt) was unclassified due to a poor signal and clustering (not shown in Fig. 4). CY063 had nearly equal proportions of G and D subtypes. It is significant that the crossover sites of the CY063 isolate recombinant form corresponds to the genomic regions (the 5′-end of RT, 3′-end of RNase and IN of pol, vpr, 5′-end of gp120, 3′-end of gp120, and 3′-end of gp41 of env) of HIV-1, which have been characterized as recombination hot spots. 29 –31 A BLAST search was performed to carry out comparative phylogenetic analysis with HIV-1 sequences from the Los Alamos HIV Sequence Database. No similar recombinant patterns were found in samples from the database, supporting the belief that this recombinant sequence is a unique recombinant form. A previous study referred to the existence of a D/G/D recombinant sequence of the protease and RT region (2042–4261 nt) of pol from a Cameroonian isolate, 32 but did not have the same recombinant pattern as the one found in this study, since CY063 has a G/D/G pattern for the same region. Additionally, one Portuguese strain was found to have a envG-nefD/G proviral genomic structure, 33 but did not match with the recombinant CY063, since it has the envD/U-nefD/U pattern, where U represents unclassified.
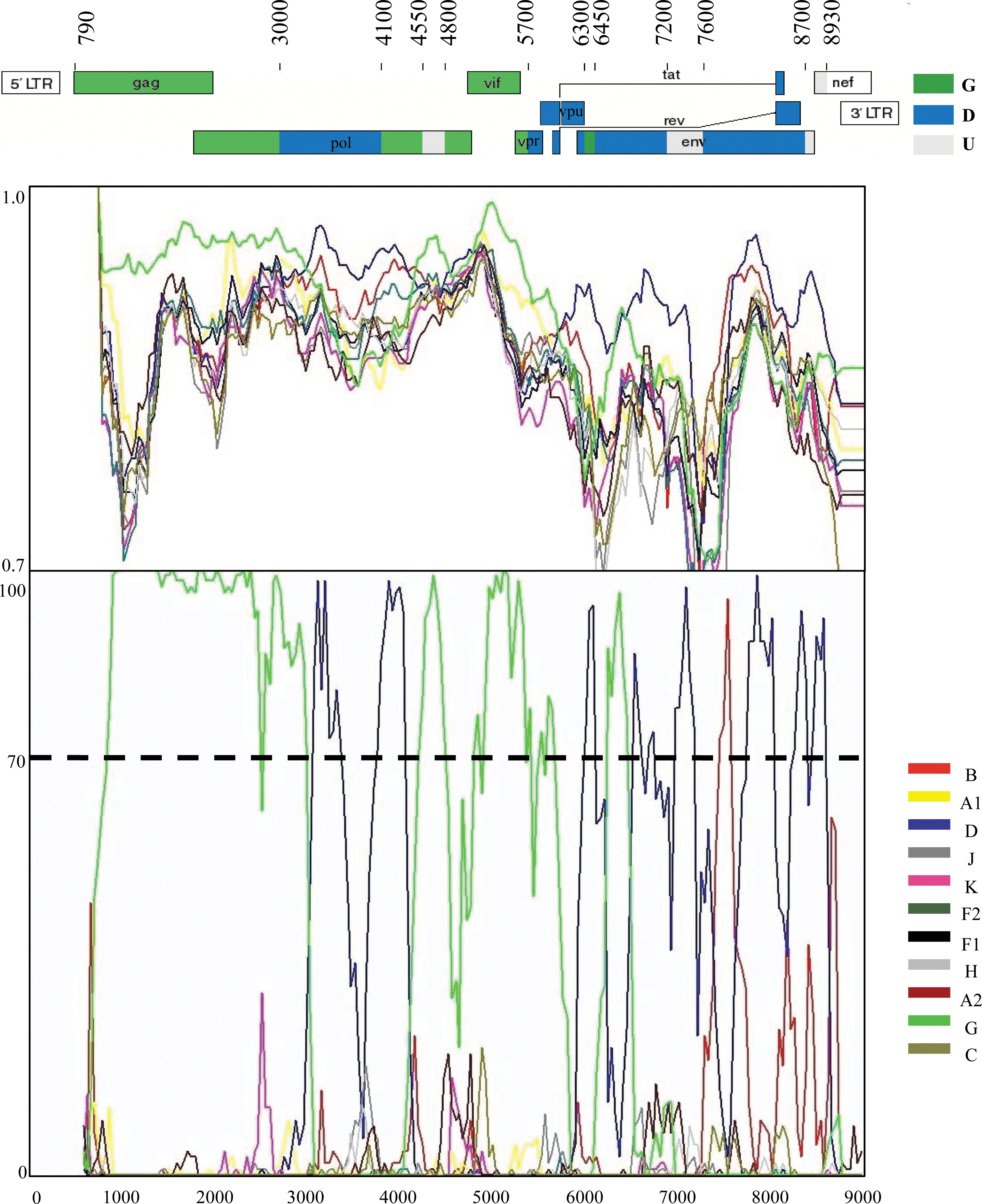
Recombinant analysis of the near full-length genome of the CY063 isolate in comparison to reference strains of all HIV-1 pure subtypes of the M group. The upper diagram indicates the gene regions and the recombination breakpoints of the CY063 isolate as determined by informative analysis. The illustration was created according to HXB2 numbering using the Recombinant Drawing tool available on the Los Alamos HIV sequence Database website.28 The middle panel presents the similarity plot diagram and the bottom panel shows the bootscan analysis performed as described in Materials and Methods. The y-axis in the similarity plot indicates the percent identity of the query sequence to a set of reference sequences; in the bootscan diagram, the bootstrap value. The x-axis shows the nucleotide position of the HXB2 genome. The dotted line indicates 70% (significant) bootstrap value.
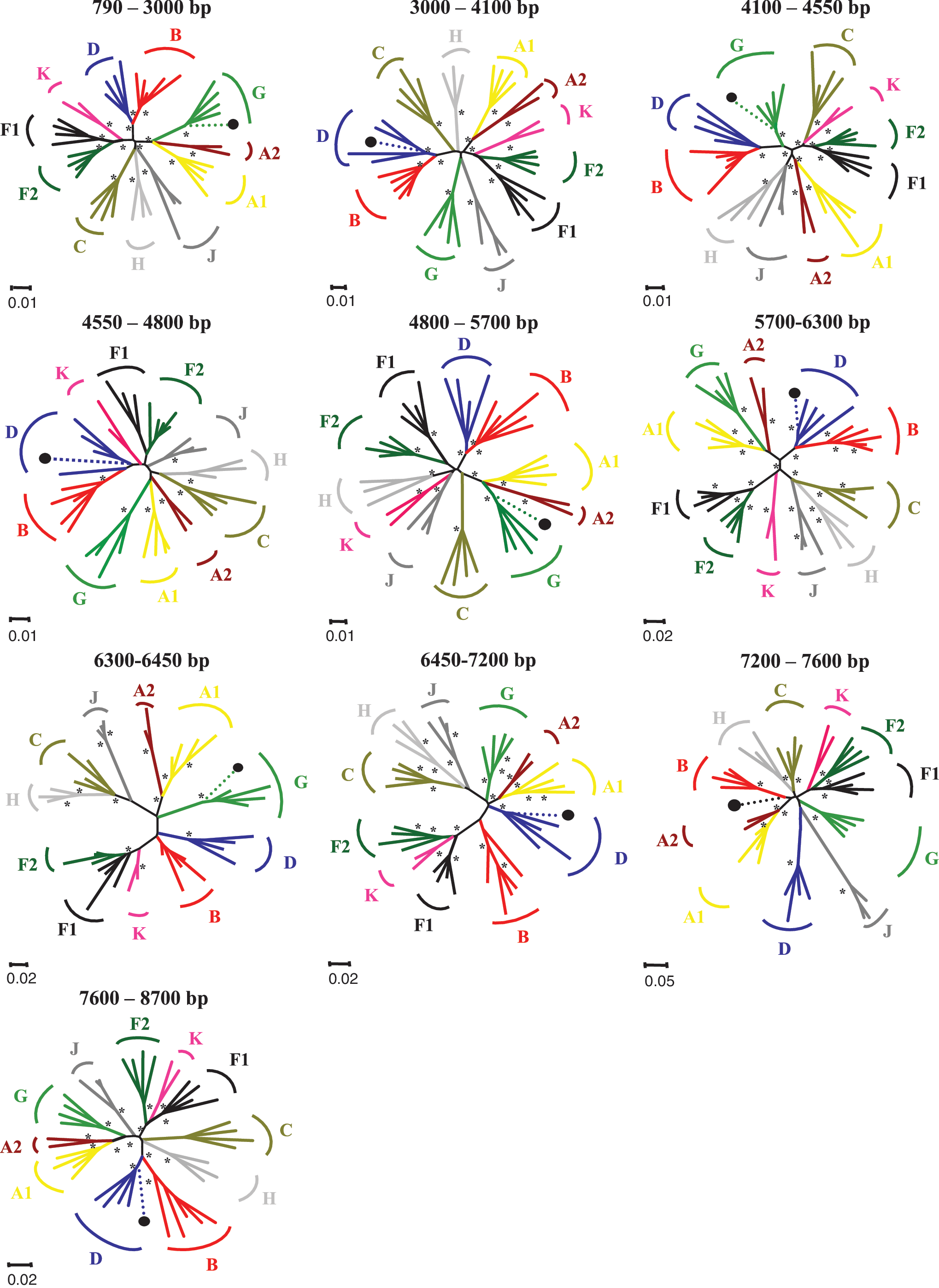
Phylogenetic analysis of the interbreakpoint segments comprises the CY063 strain as defined by the similarity plot and the bootscan analysis. Analysis was performed using the NJ method with the Kimura's two-parameter parameter distance estimation method and bootstrap analysis (1000 replicates). All reference HIV-1 pure (sub)subtypes of the M group were used to construct the trees and are denoted with different colors. *, Bootstrap value ≥70%; ∙, query sequence of CY063. Loci of genome segments are based on the HXB2 numbering. The divergence between any two sequences is obtained by summing the horizontal branch length, using the scale at the lower left.
As illustrated in the similarity plot (Fig. 5), along the CY090 sequence there are multiple regions with similarity to subsubtypes F2 or F1, but in general the similarity with these subsubtypes is not significantly higher than the similarity with the other subtypes. Further analysis of the CY090 sequence by the bootscan approach demonstrated that clustering close to subtype K existed in a few short genetic regions with bootstrap values above 70%, but in the rest of the sequence the bootstrap values with this clade were <70% (Fig. 5). A detailed similarity analysis and bootscan analysis were performed with the inclusion of near full-length sequences of all the pure subtypes and CRFs, but no difference was seen (data not shown). The limited size of the few regions that cluster with subtype K in bootscan analysis and the mixed similarity plot suggest that the new sequence does not cluster with any known subtype or CRF. An ML tree was constructed from the near full-length sequence of CY090 and all pure HIV-1 reference sequences to determine if a clustering occurs (Fig. 5). The ML tree confirms the previous analysis: it is close to subtype K, but does not totally cluster. An HIV-1 variant that belongs to the M group, with limited similarity of short genetic regions to subtype K and the remainder of the genome unrelated to any established HIV-1 subtype, was found in an HIV-1-infected male patient living in the Netherlands who most likely encountered the virus in Africa before 1989 via heterosexual contact. 34 However, phylogenetic analyses using near full-length sequences from the two unknown “K-like” strains, CY090 from Cyprus and H10986 from the Netherlands, revealed that they are unrelated HIV-1 variants (data not shown).

Recombinant analysis of the near full-length genome of the CY090 isolate in comparison to reference strains of all HIV-1 pure subtypes of the M group. The upper diagram indicates the similarity plot of the CY090 isolate; in the middle is the bootscan analysis. Similarity and bootscan plots were performed as described in Materials and Methods. The y-axis in the similarity plot indicates the percent identity of the query sequence to a set of reference sequences; in the bootscan diagram, the bootstrap value. The x-axis shows the nucleotide position of the HXB2 genome. The dotted line indicates 70% (significant) bootstrap value. At the bottom of the scheme an ML tree of the near full-length sequence of CY090 and all pure HIV-1 reference sequences and two CRFs is presented with bootstrap analysis (1000 replicates). All reference HIV-1 pure (sub)subtypes of the M group were used to construct the trees and are denoted by different colors. *, Bootstrap value ≥70%; ∙, query sequence of CY090.
Discussion
In the period 1986 to 2006, 287 persons were reported infected with HIV-1 in Cyprus. In this study, a genetic characterization and a phylogenetic analysis were performed in the near full-length genome sequences of 77 HIV-1 seropositives, depicting 38% of the known infected population of HIV-1 seropositives, to determine the genetic profile of HIV-1 strains circulating in Cyprus in the past two decades. Compared with the data already published by Kostrikis et al., 19 and Kousiappa et al., 21 the present study offers more detailed information on the molecular epidemiology of HIV-1 infection in Cyprus.
The phylogenetic analysis of the near full-length genome of the analyzed HIV-1 samples indicated clearly that subtype B is the dominant subtype, followed by subtypes A, C, and CRF02_AG, strains that dominate the global epidemic. 35 Representative strains of other subtypes have also been observed, as of subtype D, CRF01_AE, and CRF04_cpx. Two unclassified isolates derived from different HIV-1-seropositive individuals; CY063 and CY090 were further characterized phylogenetically. The CY063 isolate had a unique mosaic pattern, comprising segments of subtypes D and G and unclassified short regions. The newly found URF is probably generated from a recombination event between two parental viruses: one belonging to the subtype G lineage and the other to subtype D, in DRC where the infection was contracted. The profile of HIV-1 in DRC is characterized by high HIV-1 genetic diversity with a large number of cocirculating HIV-1 subtypes, recombinant viruses, and unclassified strains. 36,37 Subtypes D and G are present in DRC, with a regional range of 5.1–17.4% in 2002 and 6.7–13.1% in 1997 for subtype D and 2.3–21.7% in 2002 and 3.3–9.7% in 1997 for subtype G. 37 This new and unique recombinant form of HIV-1 displays a recombinant structure distinct from any other CRFs or URFs reported so far. The detailed phylogenetic analysis of the near full-length sequence of the CY090 isolate revealed that is close to subtype K, but does not totally cluster within. Similarity plot and bootscan analyses revealed that most of the genome sequence of the CY090 isolate is not closely related to subtype K sequences or any other known HIV sequence (Fig. 5). In a previous study of Kostrikis et al., 19 an isolate of patient CY090 (H044) was collected, amplified in the env (C2–V3) region, and analyzed by heteroduplex mobility assay (HMA), but could not be classified into any HIV-1 subtype (A to F) known at that time. A phylogenetic analysis with HIV-1 pure reference subtypes denoted a relation to subtype F reference sequences, the ancestor lineage of subtype K, 38 but formed a distinct divergent cluster; because it was not clear whether that sequence belonged to a new subtype or was recombinant, it was referred to as FcY. Nevertheless, although the near full-length sequence of this strain has been analyzed, no clarification about its phylogeny could be made. The CY090 strain may represent a novel subtype or a subsubtype of subtype K. At least two epidemiologically unrelated patients with an HIV-1 variant that is similar to the CY090 isolate are required to formally designate this variant as a new HIV-1 subtype or subsubtype.
The presence of a new URF and an HIV-1 diverse unclassified strain increases the genetic complexity of the HIV-1 epidemic in Cyprus. This is not consistent with the country's small area, population size, and transmission type, but it can be explained by the population flow in Cyprus of many foreign tourists, workers, political refugees, as well as repatriating Cypriots. The majority of non-B subtypes entering Cyprus in the past two decades is due to a large number of immigrants from Africa and eastern European countries, where non-B strain are predominant. 35 It is well known that Africa has the greatest genetic diversity of HIV-1, 39,40 and in this study 12.9% of the HIV-1 infections are linked to Africa, including the strains CY063 and CY090. This phenomenon has also been observed in other European countries, 41 –47 in countries of the Mediterranean region, 48,49 and in the United States. 50
Current subtype screening assays may not be sufficiently effective to capture the increasing complexity of recombinant genomes or new variants. In our method we employed near full-length characterization, a highly accurate assay that enables us to determine subtype distribution. Nevertheless, the assay must be continually evaluated and optimized in relation to newly identified recombinant forms as new and strange variants are circulating in Cyprus. 51
In conclusion, this is the largest study of phylogenetic analysis of near full-length coding sequences of Cypriot HIV-1 strains confirming the high genetic diversity of HIV-1 in Cyprus, which derives from the introduction of multiple non-B genetic forms from other countries. Furthermore, a novel URF and an unclassified variant, both originating from the DRC in Africa, were identified as part of this study. Ongoing surveillance of HIV-1 subtypes and recombinant forms in Cyprus may have important implications for HIV-1 vaccine development and new drug design and can provide insights into the evolutionary history of HIV-1.
Sequence Data
GenBank accession numbers for the near full-length sequences obtained in this study are as follows: FJ388890–FJ388965 and FJ403482.
Footnotes
Acknowledgments
We thank all participating subjects from the Larnaca General Hospital AIDS Clinic, I. Demetriades, E. Lazarou, C. Kasapis, I. Christodoulou, M. Christophina, the Cyprus Ministry of Health, and the Cyprus National Bioethics Committee for valuable assistance, as well as S. Gilliland and E. Yiacoumi for data preparation. This work was supported by grants from the European Commission (FP6-014822, QLK2-CT-2001-01344, and LSHP-CT-2006-518211), the Cyprus Research Promotion Foundation (Health/0104/22), the University of Cyprus (8037-3/312-25004 and 837-25011), and the Birch Biomedical Research LLC (3416-25017) awarded to L.G. Kostrikis.
Disclosure Statement
No competing financial interests exist.