
Abstract
The South American HIV-1 epidemic is characterized by the cocirculation of subtype B and BF recombinant variants. Together with the B and BF genotypes, HIV-1 subtype C (HIV-1C), F1, and several other recombinants have been reported. The epidemiological significance and immune correlates of these “non-B-non-BF” strains circulating in South America are still uncertain and therefore are increasingly attracting the interest of the scientific community. In this study, the South American HIV-1C epidemic was studied using new technologies for the phylogenetic analysis of large datasets. Our results indicate that there is a major clade encompassing most of the South American HIV-1C strains. These analyses also agreed that some strains do not group inside this major clade, suggesting that there could be HIV-1C sequences of different origins circulating in South America. Others have proposed different hypotheses about the origins of HIV-1C strains from South America. This study shows that an exact single origin cannot be determined, a fact that could be attributed to sampling problems, phylogenetic uncertainty, and the shortage of historical and epidemiological data. Currently, the reported data indicate that HIV-1C strains were introduced in Brazil and afterward spread to other regions of South America. By using character optimization on the obtained phylogenetic trees, we observed that Argentina could also be a point in which the HIV-1C epidemic entered South America.
Introduction
T
Subtypes and CRFs are unevenly distributed, being different subtypes largely dominant in different geographic locations. 6,8,9 All subtypes are present in sub-Saharan regions, with the highest diversity observed in Central-West Africa. Subtype C strains (HIV-1C), which dominate a geographic distribution encompassing Africa, China, and India, account for 50% of HIV infections worldwide, followed by subtypes A (12%), B (10%), G (6%), and D (3%). 9
In South America, the HIV epidemic is characterized by the cocirculation of subtype B and BF recombinants, which show a different distribution pattern across the subcontinent. 10 Subtype C and F1 infections are also important in some regions. 10 –19 Previous analyses based on sequences from the pol and vpu genes showed that small proportions of strains with subtype C, as well as BC, A2D, BD, and BK, can also be present in countries where B and BF strains are predominant. 13,17 –20 The natural history, epidemiological significance, and immune correlates of these “non-B-non-BF” strains are still uncertain. Therefore, these points are increasingly attracting the interest of the scientific community.
The HIV-1C epidemic in South America is thought to be monophyletic. 11,20 Previous studies suggest that the South American HIV-1C strains could be linked to strains from Senegal, 17 Botswana, 11 Burundi, 11 and Kenya. 21 Herein, both the monophyletic status and the relationships of HIV-1C from South America were revisited using all the previously described HIV-1C sequences from South America plus new sequences reported here and all the HIV-1C sequences available at the Los Alamos HIV dataset. The advantage of using such a large dataset is that all the known evidence (sequences) is included in our analyses. As in most scientific studies, discarding information arbitrarily can lead to unpredictable consequences for the results, many times resulting in misleading conclusions. 22 On the other hand, these studies pose an important challenge given the enormous number of possible phylogenetic trees from which to choose. 23,24 Traditional heuristic methods are useful to deal with datasets of up to ∼300 sequences. 25 For the past 20 years, methods capable of analyzing thousands of sequences have been described. Potent heuristics have been developed to use parsimony. 26,27 These methods are implemented in the TNT program, 28 which has been successfully used to analyze datasets of more than 10,000 sequences. 29 Methods for the analysis of large datasets using maximum likelihood have also been developed. 30,31 In this work, these new technologies were used to analyze our HIV-1C dataset.
Materials And Methods
Dataset
We included 51 sequences (49 from Brazil, one from Uruguay, and one from Argentina) described by Fontella et al., 21 and 12 sequences from Argentina and Uruguay previously reported, 12,13,17,19 one sequence from Venezuela 20 and four new HIV-1C sequences (198227, 176988, 146965, and 188418) identified by screening a dataset of ca. 3000 polymerase (pol) sequences from Argentina (L.R. Jones et al., unpublished results). These sequences were deposited in GenBank under accession numbers FJ659846, FJ659847, FJ659848, and FJ659849. All subtype C pol sequences available at the Los Alamos Laboratory–HIV sequence database were included in our analyses, with the exception of problematic entries. Reference sequences from the non-C subtypes were also obtained from the Los Alamos database. We used eight SIV sequences to root the trees (AF_115393, AJ_271369, AF_103818, AF_382828, AY_169968, X52154, AF_447763, and U42720). The sequences were aligned using the Mafft program 32,33 and the resulting alignment was evaluated using the Genetic Data Environment (GDE) program. 34 The sequence alignment, together with all the phylogenetic trees obtained here, are available upon request from L.R.J.
Phylogenetic analyses
Phylogenetic analyses were conducted using parsimony, 22,35 distance, 36 and maximum likelihood. 37 Parsimony analyses were performed with the TNT program 28 using a combination of Ratchet (rat), Tree Fussing (tfuse), Sectorial Searches (sectsch), and Tree Drifting (drft). 26 The maximum likelihood trees were obtained by the PhyML 30 and RaxML 31,38 programs. The MrAIC script 39 was used to infer the sequences' evolutionary model. The model to which data were best fit was HKY + I + G. PhyML was set to estimate all the model parameters during the searches. The RaxML search consisted of 1000 rapid bootstrap inferences followed by a thorough ML search. All free model parameters were estimated by RaxML. Estimating group supports in large datasets is a cumbersome enterprise, because of the prohibitive computational time required to perform regular analyses on the resampled matrices, especially with parametric methods. 29,40,41 Bootstrapped neighbor-joining (b-NJ) and a blend of symmetric resampling 42 and parsimony jackknifing 43 were used in our study. PhyML systematically crashed when asked to perform a bootstrapped analysis of our dataset. Therefore, aLRT methods 44 were used, which are relatively new approaches for branch support estimation using likelihood. The aLRT, which is a modification of the standard likelihood-ratio test, compares the likelihood of the best and the second best alternative arrangements around the branch of interest. 44 The NJ and b-NJ analyses were performed in PAUP*, 45 which was run in the Orchestra Cluster facility from the Research Information Technology Group at Harvard Medical School. The parsimony jackknife (PJ) trees were obtained by TNT.
Recombination analyses were performed by bootscanning 46 using the Simplot program 47 and the jumping alignment approach implemented in the jumping profile hidden Markov model (jpHMM) program.
Results
Herein, an analysis of sequences from the polymerase (pol) gene of 1462 HIV-1C strains is described. A total of 1409 sequences were taken from the Los Alamos database, and all the previously published HIV-1C pol sequences from South America (see Materials and Methods for details) were compiled. In addition, to identify new HIV-1C strains, a dataset of ca. 3000 polymerase (pol) sequences from Argentina obtained in our laboratory was inspected. This last analysis allowed us to identify four new HIV-1C pol sequences that were also included in the current analyses. Once aligned, our dataset was composed of 1470 nucleotide positions in the polymerase (pol) gene, of which 908 were phylogenetically informative.
Bootscanning analyses indicate that three Argentinean sequences studied here could be recombinants (Fig. 1). Two of them (146965 and 188418) were clearly C/B mosaics, whereas the third one (96105) presented a more complex genetic structure. The bootscanning assigned central regions of this last sequence to subtype D. Furthermore, this analysis suggested that the region toward the 3′ end of this sequence could be assigned to subtype B (Fig. 1). The analyses performed with jpHMM confirmed the results obtained by bootscanning for strains 146965 and 188418 and indicated that the most likely recombination breakpoints are located at the intervals 2945 ± 22—3201 ± 23 and 2946 ± 26—3199 ± 21, respectively (coordinates based on HXB2 numbering). On the other hand, these analyses indicated that the central region of strain 96105 could not be assigned to subtype D but confirmed that the 3′ end belonged to subtype B (posterior probability = 1, Fig. 1). The structure of both 146965 and 188418 was, therefore, nearly the same as the one reported for B/C recombinants described previously in Brazil. 48 –50 Conversely, the breakpoint of the 96105 sequence is probably located at position 3475 ± 9, suggesting that this strain could be a member of a different C/B CRF circulating in Argentina (Fig. 1).

Bootscanning and jpHMM analyses of newly identified recombinant sequences from Argentina. For the bootscanning analyses (
The parsimony analyses resulted in 24 equally parsimonious trees 11,881 steps long. The corresponding strict consensus tree is shown in Fig. 2. The sequence from Venezuela (hereafter the Venezuelan clade, A in Fig. 2) was closely related to strains from Zambia, forming a small clade of three sequences that was supported in the PJ tree (Fig. 3).The Argentinean strain 198227 was linked to strains from Zimbabwe, Botswana, Tanzania, and Zambia, though this clade was not supported in any of the resampled analyses. The largest clade in Fig. 2 (hereafter the South American, SAM, clade) encompasses all strains from Brazil, as well as all sequences from Uruguay and most of the Argentinean strains. This is an expected observation when we consider the strong social and commercial links that exist among these countries. Equivalent results were obtained by NJ and maximum likelihood analyses (data not shown). Though the majority of strains were clustered in a single clade, the HIV-1C strains from South America are not monophyletic but are distributed along three independent, distantly related sections of the subtype C clade. These findings are in contrast to those previously reported that suggested that South American HIV-1C is monophyletic.
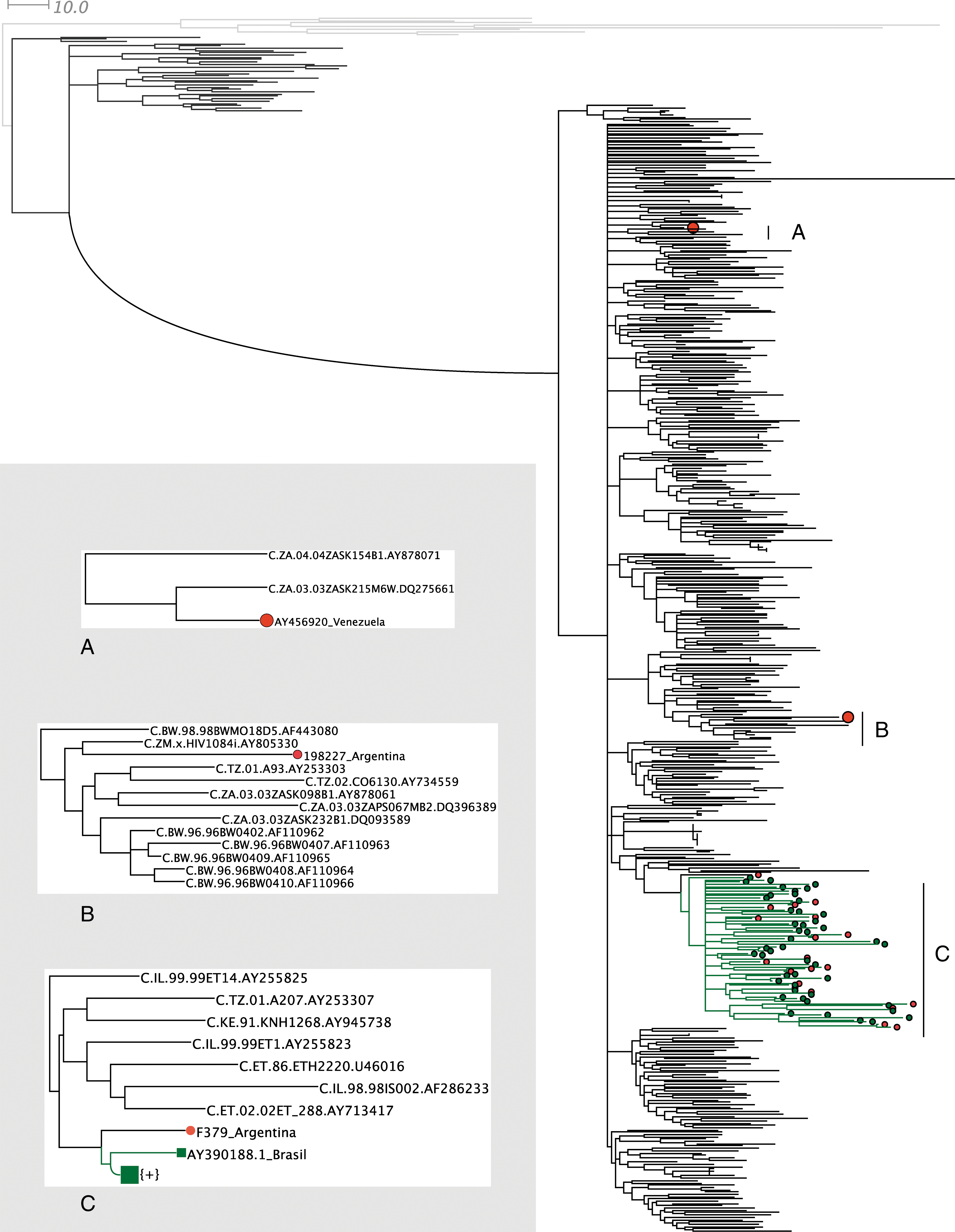
Strict consensus tree representation of the 24 most parsimonious trees (L = 11,881) obtained using the new technologies implemented in the TNT program. The outgroup branches are colored light (SIV) and dark (non-C subtypes) gray. The South American sequences are indicated in green (Brazilian) and red (Argentinean, Venezuelan) circles. The inset displays in detail the three regions of the HIV-1C clade (

Parsimony jackknifing tree. Outgroup sequences are indicated with bold (SIV) and gray (non-C subtypes) branches. The larger cluster, showing a support of 100, corresponds to the HIV-1C clade. Inside this cluster, the SAM clade has a support of 56, the Indian clade of 68, and the Venezuelan clade is weakly supported with a value of 51. The South American terminals are indicated by black circles (Argentinean and Venezuelan strains) or gray squares (Brazilian strains). The Indian clade is indicated by gray circles.
Resampling analyses displayed the same major structure of well-supported nodes, with the HIV subtypes supported by high bootstrap, jackknife, and aLTR values (data not shown). The PJ tree in Fig. 3 displays jackknife values for HIV-1, HIV-1C, and subclades inside HIV-1C. The Venezuelan clade was present only in the PJ tree and was weakly supported. The SAM clade was also weakly supported in the PJ tree and was absent in the b-NJ analyses. Nevertheless, it was strongly supported in the aLTR analyses (aLTR: 0.95; SH-l: 0.94; Chi: 0.99). The linkage of the Venezuelan strain with sequences from Zambia, as well as the clade containing the 198227 Argentinean strain, was unsupported in both the b-NJ and aLTR trees. The previously recognized Indian clade 54 –56 also showed low support values (Fig. 3; b-NJ: 78; aLTR: 0.76; SH-l: absent), indicating that the support estimates observed here are quite conservative.
Phylogenetic trees can be used to infer the most likely geographic origin of a given clade. Furthermore, it has been shown that the pol gene is useful for the identification of transmission events by phylogenetic means. 57,58 The geographic origin of a clade can be tracked by traveling downward in a rooted tree to the node that connects the clade of interest with its sister group (Fig. 4).Sometimes, complications can arise due to (1) phylogenetic uncertainty and (2) the configuration of the sister clade. The first point refers to the fact that it is possible to have a set of equally good trees with different sister groups for the test clade (Fig. 4a and 4b). The second one is given by the fact that the sister clade can be composed of taxa from more than one origin (Fig. 4c). Here, we observed both phylogenetic uncertainty and topological difficulties when trying to identify the most likely origin of the South American HIV-1C strains. In addition to this problem, sample bias and the natural movement of strains between neighbor regions could negatively affect the interpretation of phylogenetic trees. For instance, finding HIV-1 in Zimbabwe somewhat related to HIV-1 in Brazil just as likely is the result of strains moving from Ethiopia to both Zimbabwe and Brazil as it is the result of HIV-1 evolving in Zimbabwe and then migrating to Brazil. To mitigate, at least in part, these problems we down-navigated the trees up to three nodes toward the root from the three taxa of interest (Venezuelan, 198227, and SAM) and annotated the origin of all the strains derived from those nodes. The results of these analyses are summarized in Table 1. From these results, it is clear that several countries from the East-Center of Africa could be the origin of the South American HIV-1C strains. Whether the inability to identify a single origin for South American HIV-1C is due to phylogenetic uncertainty or incomplete sampling remains to be investigated.
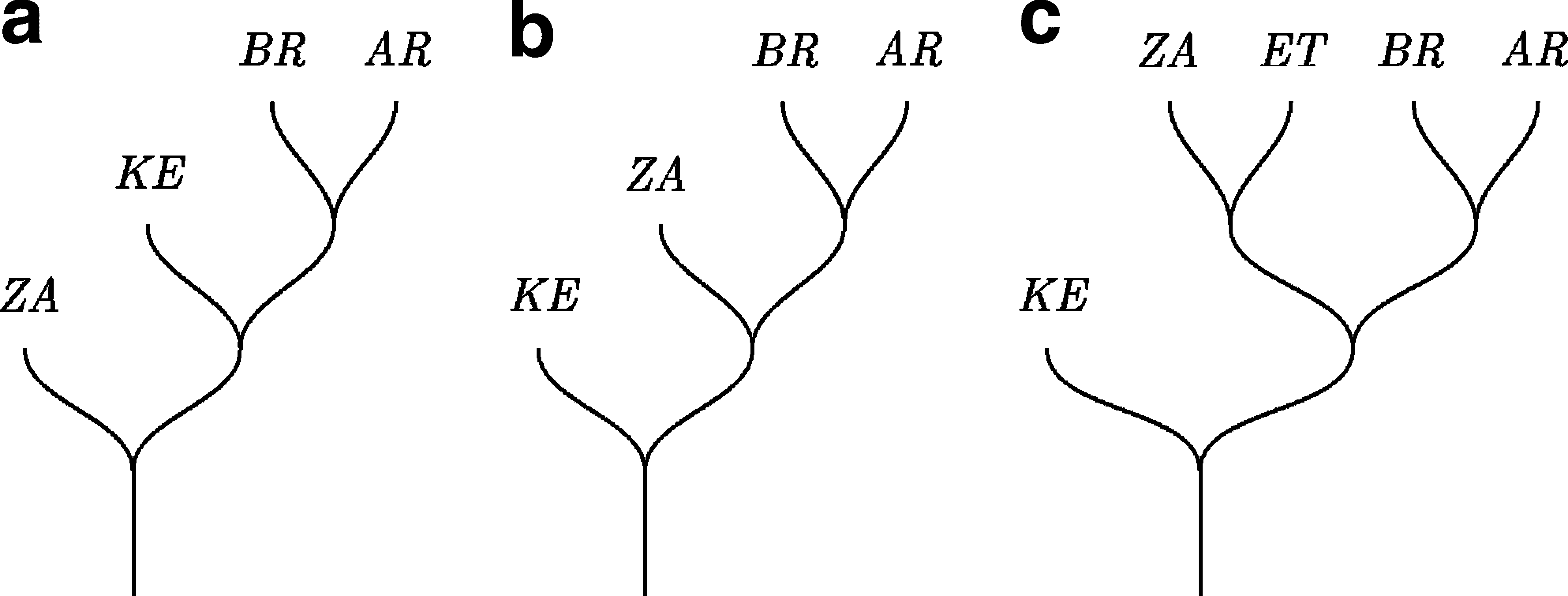
Toy phylogenetic trees exemplifying different possible scenarios that could be observed during the inference of the geographic origin of a given clade [in our example, the group (BR, AR)]. The terminal's names indicate geographic locations. In (
Supported (>50%). One to three asterisks are shown in the PhyML analyses indicating support in one, two, or three of aLTR, χ2, and SH-l analyses.
There is agreement that the SAM HIV-1C clade in South America first entered Brazil, after which the viruses spread to other countries. 10,11,13,21,59 Nevertheless, we observed that HIV-1C sequences from Argentina were interspersed inside the SAM clade and, furthermore, that an Argentinean sequence was located at the base of the SAM clade (Figs. 2 and 3). These results support the idea that Argentina may also be an entry point for the HIV-1C strains in South America. The center of origin of a clade can be studied using character optimization, which is the process used to infer the most plausible states at the tree nodes, using the evidence provided by the actual data. 25 The node states discovered by this process are considered ancestral, indicating the past characteristics of the clades under study. In our case, the state (geographic location) at the base node of the clade of interest is inferred as the most plausible origin of the clade. 60 Here, character optimization was performed by hand using the Fitch optimization algorithm for unordered multistate characters. 61 This procedure was applied in each of the parsimony trees and the trees obtained with PhyML, PAUP*, and RaxML. The results are summarized in Table 2. In the NJ and PhyML trees, “Brazil” was the most plausible state of the base node of the SAM clade. In the parsimony and RaxML trees the optimization was ambiguous (Argentina/Brazil), suggesting that Argentina or Brazil may be the ancestral state of the SAM clade.
Four nodes are shown, starting from the base of the clade of interest toward the root.
The states in all optimal trees were pooled.
Base node of the SAM clade.
Discussion
The introduction of HIV-1 strains into new populations, countries, and regions, in association with social and cultural aspects of humankind, indicates a dynamic scenario for the evolution of HIV. An important induction derived from the analyses described here is that HIV-1C strains from South America have a polyphyletic origin (Figs. 2 and 3). However, most of the strains grouped into the monophyletic SAM clade described previously, 11,21 suggesting that the largest part of the epidemic is monophyletic. It is difficult to speculate on the size of the South American HIV-1C epidemic. As part of an ongoing analysis we have observed that 8 out of 2906 strains from Argentina presented sequences of subtype C. It is thought that the number of people living with HIV in Argentina is ∼120,0005; thus, we could determine that a rough estimate of the number of HIV-1C-infected individuals is about 300. In Venezuela, the estimated number of infected people is 170,000. 20 In a study published in 2005, Castro et al. 20 observed that 2 (1 pure, one recombinant) out of 106 cases from Venezuela had C subtype sequences, suggesting a proportion of HIV-1C larger than the one observed in Argentina by one order of magnitude. In a study performed in Peru, Yabar et al. 62 observed that 1 out of 19 randomly taken samples was a B/C recombinant, also suggesting the circulation of HIV-1C strains in this country. Thus, our sample might be too small to yield conclusive results. We think that if the whole epidemic were monophyletic, the probability of observing sequences unrelated to the major clade would be very low. Therefore, the fact that we observed sequences from Venezuela and Argentina that were unrelated to the SAM clade suggests that HIV-1C sequences of uneven origins circulating in South America might exist. In addition to the fact that the number of known HIV-1C sequences from South America is small, we lack sequence data from many regions of South America. Only studies based on much larger amounts of South American HIV-1C sequences, obtained from a wider geographic range, will allow us to determine the epidemic in depth.
The precise origin of the South American HIV-1C strains remains elusive (Table 1). Neither previous evidence of a Senegalese origin of some subtype C strains circulating in Argentina 17 nor the Burundian link observed for Venezuelan sequences 11 could be supported by the present analyses. Our results do not contradict the previous hypothesis linking the South American strains with sequences from Botswana 11 and Kenya, 21 though these hypotheses were neither univocally supported (Table 1). Our analyses agreed that the origin of the South American HIV-1C strains could be located somewhere in Middle-Eastern Africa. Nevertheless, these results must be interpreted with caution, for sampling of HIV-1C in Africa is highly biased and nonrandom and, as discussed in the Results section, the likely introduction of subtype C into South America happened many years ago and, at the same time, subtype C was moving around Africa.
Previous epidemiological data support the hypothesis that Brazil is the center of dispersion of the large monophyletic HIV-1C clade in South America. 10,11,13,21,59 Nevertheless, the analyses performed in this study suggested the possibility that the center of dispersion could be located as well in Argentina (Table 2). Most sequences from this clade have a Brazilian origin (49/68), a fact that might bias the results of the optimization procedure toward Brazil. Thus, more studies are required that include a broader sample of sequences from Argentina, Uruguay, and other neighbors to accurately infer the center of dispersion of HIV-1C in South America.
The recombination analyses performed here showed that the mosaic structure of two C/B recombinants from Argentina, which were sampled in 2004 and 2006, is almost the same as the structure of a B/C CRF reported for Brazil in 2006, 50 showing that they correspond to independent isolates of the CRF31_BC, which apparently was circulating in Argentina 2 years before its identification in Brazil. Likewise, the jpHMM analyses demonstrated that the third C/B recombinant described here has a genomic structure that is not the same as the one present in the Brazilian C/B CRF. This sequence was obtained in the year 2000, suggesting that a CRF different from CRF31_BC was circulating in Argentina before the circulation of CRF31_BC in Brazil. Unfortunately, this is the only known sequence presenting this structure, so we cannot be sure whether it represents a CRF or a sporadic recombinant strain.
Conclusions
Although the major epidemic seems to be monophyletic, HIV-1C strains from South America are polyphyletic. Our phylogenetic analyses support the idea that at least three introductions of distantly related strains guided the distribution of HIV-1C in South America. The HIV-1C epidemic in Venezuela could have originated from an introduction of strains from Zambia or Botswana, though the strains were also linked with sequences from Yemen, Zimbabwe, and Kenya in two of the four analyses performed here. The origin of the 198227 sequence from Argentina, which clustered outside the SAM clade, is obscure: it could be related to strains from Zambia, Botswana, Tanzania, or Malawi. The SAM clade origin is also elusive. Altogether, these results suggest that analysis of viral sequences alone is highly unlikely to be able to reconstruct the true epidemiology and, thus, in addition to the need or using larger numbers of South American HIV-1C sequences, as discussed above, the addition of historical information as well as more epidemiological data is a “must” for future studies.
Footnotes
Acknowledgments
Continuous support from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) is greatly appreciated. We thank Elizabeth Stansell for valuable comments and proofreading of the manuscript. L.R.J. and J.M.M. thank the Research Information Technology Group (Harvard Medical School) for granting access to the Orchestra cluster facility. The comments and suggestions of Dr. Brian Foley greatly improved our analysis and the manuscript, and are much acknowledged. We are indebted to Professor Sergio Mazzini for proofreading the manuscript.
Disclosure Statement
No competing financial interests exist.