
Abstract
In previous studies on mechanisms of HIV-1-mediated pathogenesis we showed that bystander apoptosis mediated by cell surface-expressed HIV-1 Env correlated with the fusogenic properties of the gp41 subunit of Env. A crucial step in HIV gp41-mediated fusion is the refolding of the protein into a six-helix bundle along the N- and C-terminal coiled-coil domains. These domains have been targeted by peptide inhibitors that inhibit gp41-mediated fusion. One of these inhibitors, enfuvirtide, is the first such drug approved for therapy. More recently, clinical data suggest that the beneficial effects of enfuvirtide extend beyond virus suppression and are associated with certain resistance mutations in gp41. In this study we characterized the bystander apoptosis-inducing potential of mutants associated with increased CD4 counts that arise during enfuvirtide therapy. Whereas all mutant clones were reduced in both cell-to-cell fusion activity and apoptosis induction there was limited effect on virus infection or replication. The viruses were found to have apoptosis-inducing activity in the order WT > V38M > V38A > G36D > V38E, which correlated with cell-to-cell fusion but not infection. Interestingly, the level of resistance as determined by the IC50 of enfuvirtide also correlated inversely with both cell fusion and apoptosis in that the most resistant Envs were the least fusogenic and pathogenic. This suggests the beneficial effects of enfuvirtide therapy beyond virus suppression may be mediated by selecting less pathogenic HIV isolates over time.
HIV
The Env glycoprotein is composed of a gp120 subunit, which engages chemokine receptors (CXCR4/CCR5) after binding to CD4, and the gp41 subunit, which mediates fusion of viral and cellular membranes in a pH-independent manner. 10 HIV gp41 is a classic type 1 fusion protein that contains two fairly conserved coiled-coil domains referred to as C- and N-terminal heptad repeats (HR1 and HR2). These regions interact with each other in a leucine zipper-like fashion to mediate membrane fusion. 11 This property of gp41 has been exploited in the development of peptides that mimic the heptad repeats and prevent fusion. 12 Enfuvirtide is the first such peptide approved for clinical use in HIV salvage therapy.
The immunological benefits of enfuvirtide therapy have been demonstrated beyond virus suppression. 13 It has been shown that enfuvirtide inhibits bystander apoptosis induced by Env glycoprotein both in vitro 14,15 and in vivo. 16 Furthermore, recent clinical findings suggest that certain enfuvirtide-resistant mutants arising during salvage therapy are associated with an increase in CD4 counts even after virological failure. 17,18 More specifically, mutations associated with the V38 positions were found to be associated with this phenomenon. 17 –19 Enfuvirtide resistance is known to be associated with a highly conserved domain in the N-terminal HR-1 of gp41 spanning amino acids 36 to 38 (GIV) and the majority of mutations arising during therapy are found within this region. 20,21 It is unclear as to what effect these mutations have on HIV pathogenesis, especially pertaining to Env-mediated bystander apoptosis induction.
Using a mutational approach we have previously demonstrated that the bystander apoptosis mediated by HIV Env glycoprotein is gp41 function dependent. 5 We have also shown that the single-cell apoptosis-inducing potential of Env is dependent on the cell-to-cell hemifusion-inducing potential of gp41. Furthermore, the enfuvirtide-resistant virus G36D (G547D based on Env numbering) was shown to be reduced in its bystander apoptosis-inducing potential. However, it is unclear whether the increase in CD4 counts seen in patients with viruses harboring V38 mutations is a result of reduced pathogenesis and bystander apoptosis induction. The objective of the current study was to define the apoptogenic potential of common enfuvirtide-resistant mutants arising during therapy and determine whether a correlation exists between the beneficial effects of certain mutants after virological failure and bystander apoptosis induction.
We introduced specific enfuvirtide resistance mutations in the gp41 region of our Lai Env construct and examined the effect these mutations have on bystander apoptosis induction, cell-to-cell fusion, and virus infection. HeLa cells transiently transfected with different Env constructs were cocultured with either SupT1 cells for apoptosis detection or TZMbl cells for the determination of cell-to-cell fusion. As seen in Fig. 1A, most of the enfuvirtide-resistant mutants were reduced in apoptosis induction as well as cell-to-cell fusion activity (Fig. 1B). The mutants showed a range of activity with V38M showing the greatest apoptosis and fusion activity similar to wild type (WT) and V38E showing the least apoptosis-inducing activity and being the most different from WT. The order of apoptosis induction was WT > V38M > V38A >G36D > V38E. Similar results were seen with cell-to-cell fusion experiments. On the other hand, virus infection (using single round pseudotyped infection experiments) was not severely affected by any of the mutants (Fig. 1C), with most mutants showing at least 50% infectivity of WT virus. None of the mutants had any defect in Env expression and processing (Fig. 1D) or Env incorporation in virions (Fig. 1E). This suggests that although the mutants retain their virus infection capacity they are altered in bystander apoptosis and fusogenic activity. Our study supports the idea that the increases in CD4 count may be a result of this reduced apoptosis-inducing activity of some enfuvirtide-resistant mutants.
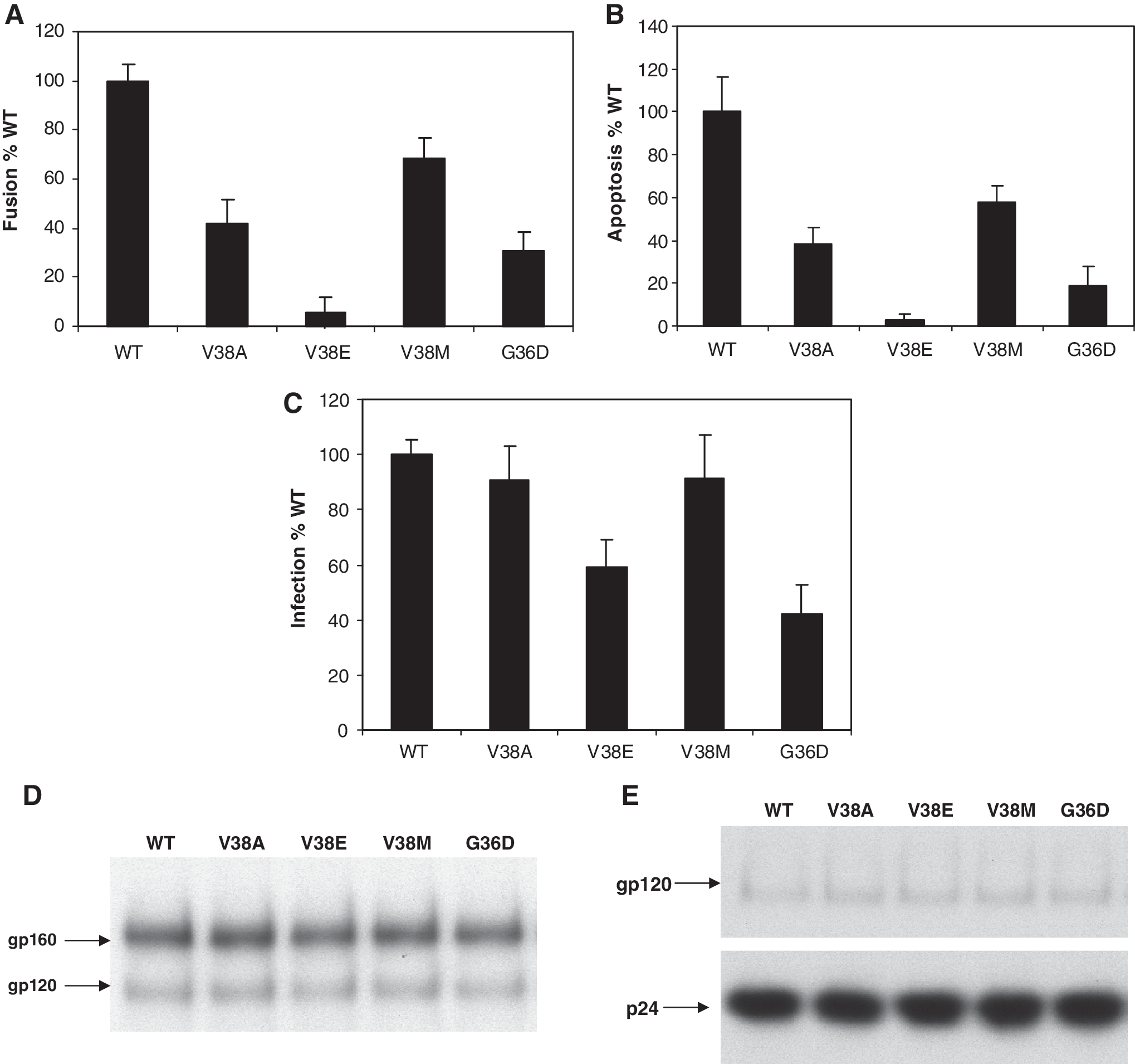
(
Having observed the altered apoptosis-inducing activity in bystander cells by these mutants without having a significant effect on virus infection we wished to determine the mechanism by which some mutants were more fusogenic than others. It should be kept in mind that the sequence of enfuvirtide is identical to the HR2 domain of gp41 and hence can bind HR1 in a manner similar to HR2. Hence, we wished to determine the degree of resistance to enfuvirtide for each mutant as the decreased fusion activity suggests that there may be an effect on the level of enfuvirtide resistance as well. Considerable variation has been observed in different mutants with regard to the degree of resistance and we speculate that more resistant viruses would be significantly different from WT virus in their interaction with enfuvirtide as well as the corresponding HR2. As seen in Fig. 2, the mutants varied considerably in the level of resistance to enfuvirtide in a pseudotyped virus infection assay. V38E was found to be the most resistant with an IC50 of 2097.84 ± 60.54 nM whereas V38M was the least resistance with an IC50 of 46.69 ±29.90 nM (Table 1). The degree of resistance was in the order WT < V38M < G36D < V38A < V38E (Table 1). In previous experiments we saw a similar trend with regard to apoptosis induction and cell fusion. In fact, a regression analysis of the correlation between various aspects of the mutants (Fig. 3) shows that apoptosis correlates strongly with cell fusion (R 2 =0.983) and inversely with the IC50 of enfuvirtide (R 2 = 0.9028) but poorly with virus infection (R 2 = 0.5932). Similar results in terms of fusion activity and fold increase in enfuvirtide sensitivity have been shown for other mutants. 22,23 This suggests that there may be an inverse correlation between level of enfuvirtide resistance and bystander apoptosis induction. However, the reason for the development of certain mutants over others in patients remains uncertain.
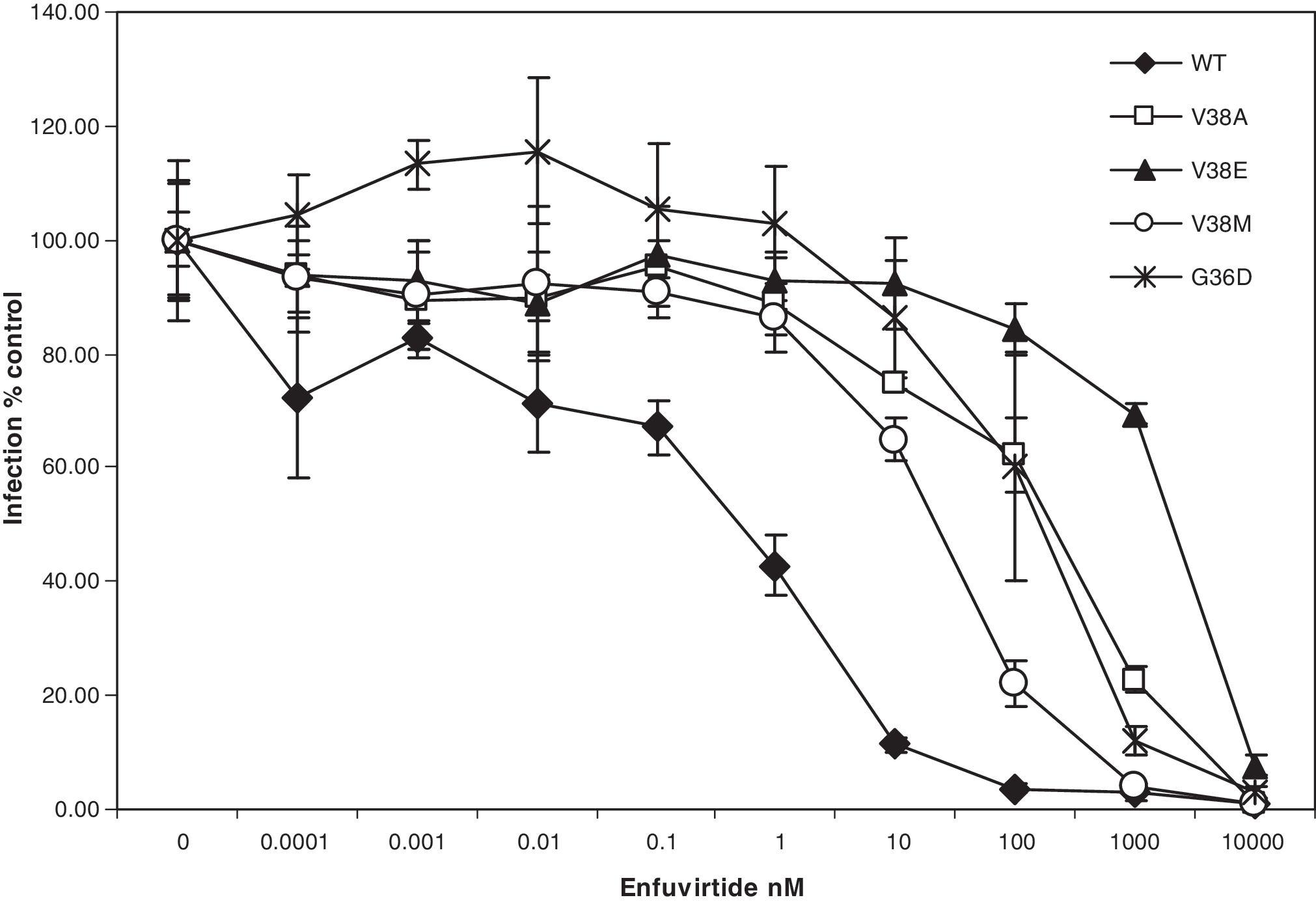
Level of resistance to the peptide inhibitor enfuvirtide using pseudotyped virus infection. The pseudotyped virus infection assay was performed in the presence of the indicated concentrations of enfuvirtide. Virus infection was determined as luciferase reporter gene readout 24 h postinfection. Data were normalized to no drug control for each mutant.
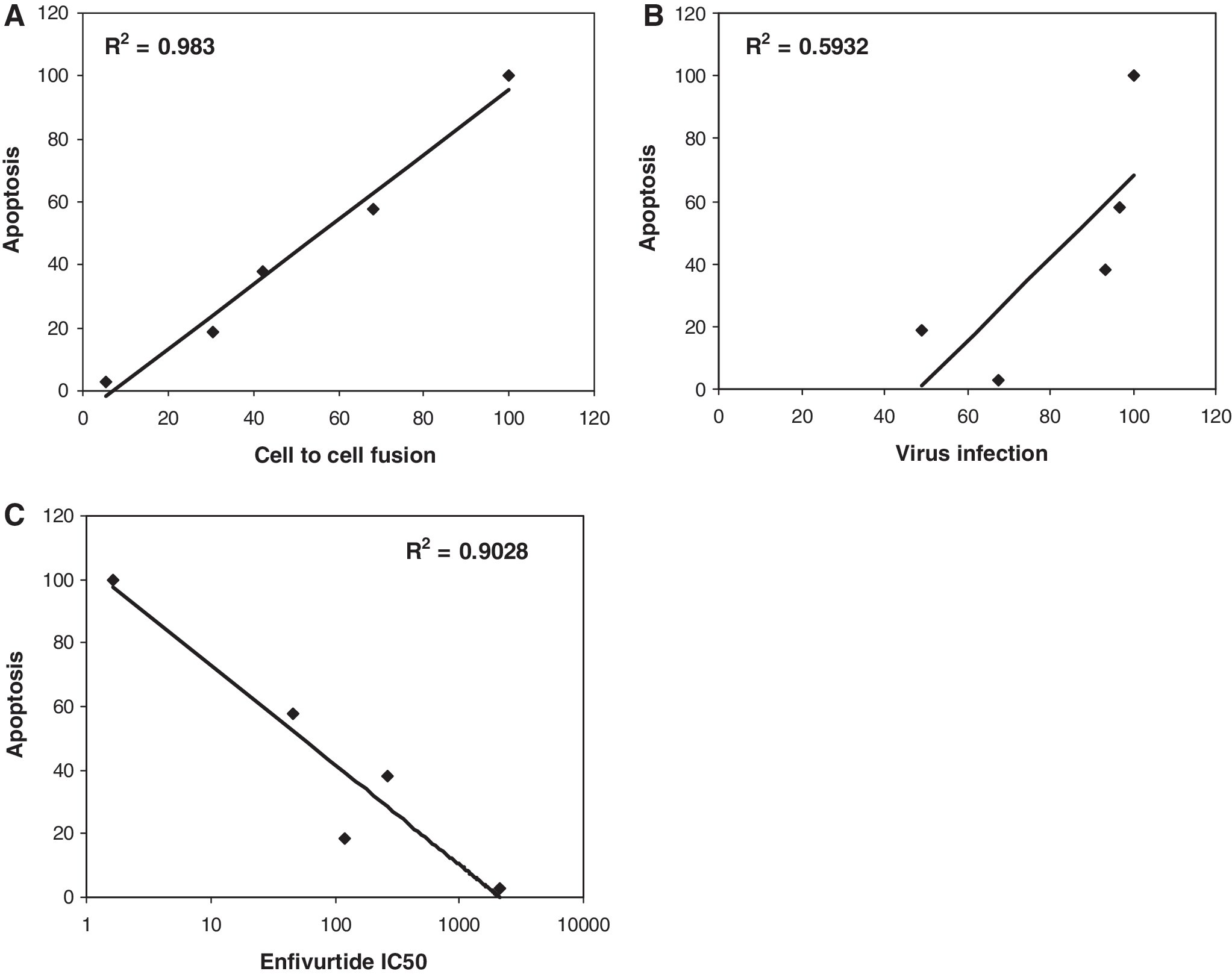
Regression analysis of the correlation between apoptosis and cell fusion (
IC50 was calculated by fitting curves using sigma plot software. Data represent averages from three independent experiments ±standard deviation (SD).
WT virus represents Env from the LAI molecular clone. All other mutations were introduced in the LAI Env backbone using site-directed mutagenesis.
We previously reported that the kinetics of replication of virus showed an inverse correlation to bystander apoptosis. 5 This is likely due to the induction of apoptosis in bystander cells by more fusogenic viruses, thereby reducing the availability of target cells. Env glycoprotein that is defective in cell-to-cell fusion but capable of virus infection (W596M) has been shown to replicate faster than WT virus. 5 In the case of enfuvirtide-resistant viruses, we also saw that although cell-to-cell fusion was severely affected, especially for mutant V38E, the virus was still capable of infection. Hence, we tested the virus replication kinetics of enfuvirtide-resistant mutants in an SupT1 cell line to determine if there was a correlation with bystander apoptosis. As seen in Fig. 4, V38E and V38A along with G36D replicated faster and to higher levels than WT. On the other hand, the replication kinetics of V38M seemed to be strikingly similar to WT, consistent with its apoptosis-inducing capacity and decreased CD4 counts in vivo. This suggests that viruses that were poor inducers of bystander apoptosis do in fact replicate faster and better than WT in SupT1 cells. Furthermore, a threshold level of virus infection (Env function) seems to be sufficient for virus replication but not for apoptosis induction. These results further support the argument that V38A/E and G36D mutants may be less pathogenic in vivo.
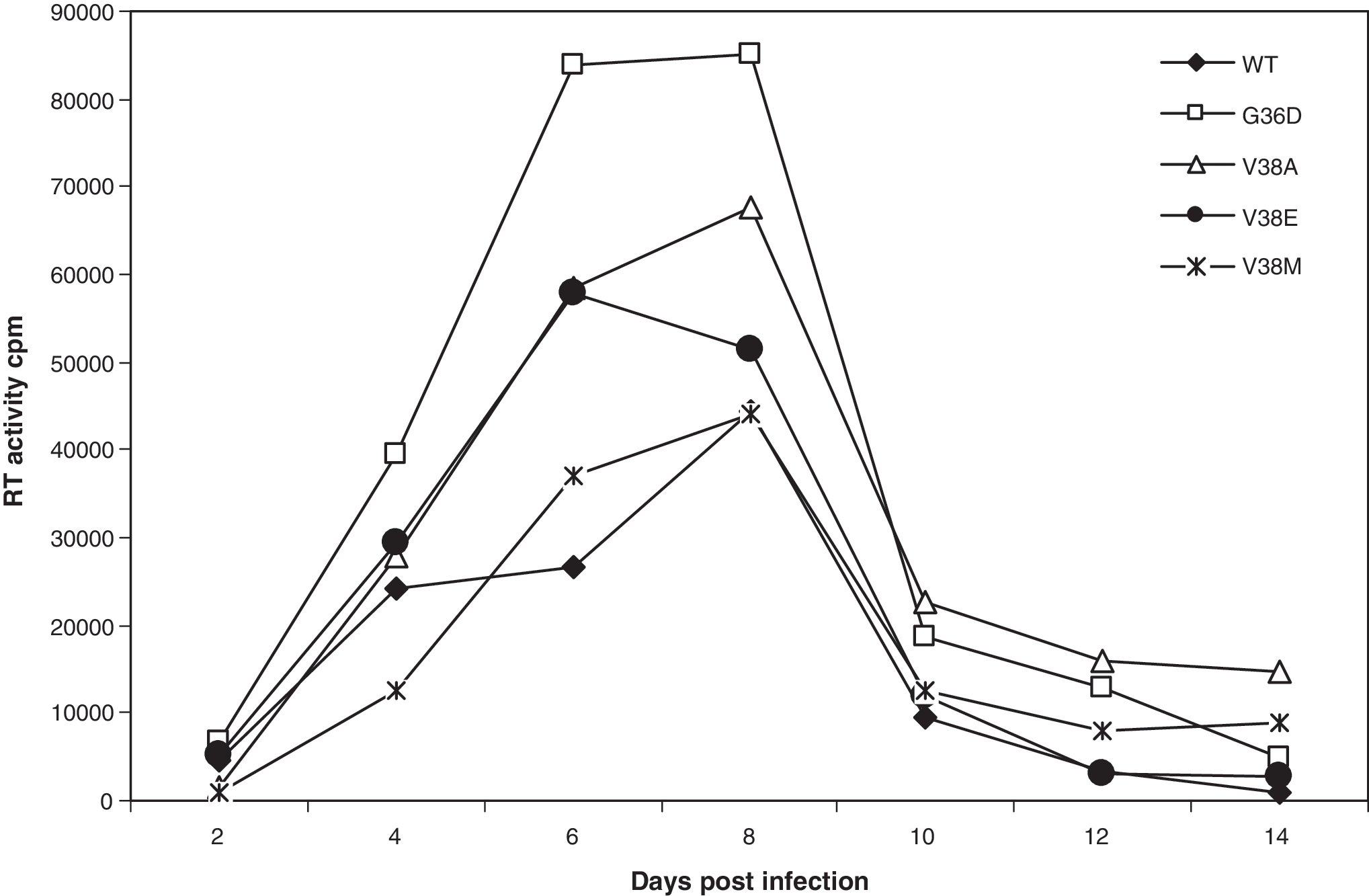
Replication of various Env mutant viruses in SupT1 cells. Equal RT values of different viruses were used to infect SupT1 cells. Supernatants were collected at indicated time points and analyzed for virus replication by RT assay. One of two representative experiments is shown.
Our findings are largely in agreement with the hypothesis that enfuvirtide-resistant mutants have lower bystander apoptosis induction and cell-to-cell fusion activity. We specifically looked at mutations arising in the conserved GIV domain spanning amino acids 36–38 of gp41. We find that one such mutant, V38E, was the poorest inducer of apoptosis, whereas V38M was capable of apoptosis induction in almost 60% of WT. Furthermore, the replication kinetics of V38M was also similar to WT. This is an interesting observation as V38M has been shown to be associated negatively with CD4 counts in patients. 17 The other mutants, V38A and G36D, were found to be intermediate inducers of apoptosis in our experiments. Although V38A/E have been associated with increased CD4 counts in a study by Aquaro et al., 17 the increase in counts associated with G36D was not statistically significant. However, in another study by Descamps et al., 24 G36D was associated with increased CD4 counts, although this was not statistically significant. This variability in clinical studies underscores the complex nature of HIV Env glycoprotein and the rather large diversity within a patient group probably due to the existence of quasispecies and baseline polymorphism in both gp41 and gp120. 25 Baseline polymorphism can severely affect the resistance pattern to enfuvirtide 26 and secondary or compensatory mutations in the HR2 27 domain of gp41 in patients may affect the fusion activity in different ways, which is not reflected in our study. Furthermore, Env fusion activity is also in part dependent on the gp120 phenotype. 28,29 Studies have shown that certain gp120 have higher CD4 or coreceptor affinity, in turn altering fusion activity 30 and pathogenesis.
Nevertheless, in our model system in which point mutations are made in gp41, a more controlled assessment can be made as to the effect of individual mutations on the virus phenotype. The resistance level seen with individual mutation has been shown to result in a direct lowering of binding affinity for enfuvirtide. 21 This means that the corresponding N-terminal heptad repeat HR1 would also have reduced affinity for the corresponding C-terminal repeat HR2. A reduced affinity between the HR1 and HR2 domains would result in reduced stability of the six-helix bundle, which has been associated with reduced fusion activity. 22 It is thus not surprising that the mutant with the most resistance to enfuvirtide (V38E) was the poorest inducer of cell-to-cell fusion and apoptosis. However, virus infection was less severely affected with single round infections, showing almost 70% infection compared to WT. Although the basic mechanisms underlying HIV-1 Env-mediated virus–cell and cell–cell fusion may be similar, 10 the differences between the two are likely to be due to the relatively small quantity of Env required for successful viral fusion 31 compared to a large number of Env reactions required for cell-to-cell fusion 32 and likely for induction of apoptosis. Furthermore, it emphasizes the fact that cell-to-cell fusion activity is not required for successful infection and replication by HIV.
The reason for the selection of certain mutations in patients over others remains uncertain. Our findings that increased resistance to enfuvirtide drives the virus further from the WT phenotype and compromises the fusogenic activity merit further investigation. It has recently been reported that the V38E mutation is selected in vitro in response to exposure to the second-generation fusion inhibitor T1249. 33 In our assay V38E was found to be the most resistant to enfuvirtide and the least pathogenic, raising the possibility that perhaps a new generation of gp41 inhibitors may be able to select even more divergent and attenuated viruses.
Our findings provide a rational explanation for the beneficial effects of certain enfuvirtide-resistant mutants arising in vivo. It also raises several important questions such as what factors are responsible for the selection of one mutant over the other, whether a rational therapy can be designed to select low fusion-inducing mutants, and whether other inhibitors targeting HIV Env would also result in an attenuated HIV phenotype. Further analysis using clinical isolates directly from patients undergoing enfuvirtide therapy would be required to address these specific issues. However, these initial findings provide support for the hypothesis that HIV gp41 is a critical mediator of HIV pathogenesis and it may be possible to target gp41 in order to attenuate HIV, making it a less pathogenic if not a nonpathogenic infection.
Footnotes
Acknowledgments
We thank Eric Freed for his support in performing the replication experiments and insightful comments. We are grateful to the NIH AIDS Research and Reference Reagent Program for supplying valuable reagents. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Disclosure Statement
No competing financial interests exist.