
Abstract
Vaccination is recommended for HIV-infected patients. Transient increases of viral load (VL) and risk of developing resistance to HAART have been described. In addition, VL rebounds could increase HIV-specific immune responses. Twenty-six successfully treated HIV-infected adults were randomized to receive a vaccination schedule or placebo during 12 months. Afterward, HAART was discontinued. Influences of vaccination over VL, genotypic mutations, different T cell subsets, and HIV-1-specific immune responses were evaluated. Patients did not present any secondary effect. No differences in incidence of detectable VL determinations were detected between groups [relative risk 0.54 (95% CI 0.23–1.26)]. No relevant resistance mutations were detected. The vaccinated group showed a significant drop in CD4+ T cells (p = 0.046) associated with increases in activated T cells. HIV-1-specific lymphoproliferative responses increased more in the vaccinated group during the vaccination period. Viral rebound dynamics after interrupting HAART were similar in both groups. A vaccination schedule in successfully treated HIV patients was safe, was not associated with an increase in detectable VL, and did not increase the risk of developing resistance mutations. However, it induced an increase in T cell activation and a drop in CD4+ T cells, although these changes did not influence the VL rebound dynamics after HAART interruption.
Introduction
T
On the other hand, it has been reported that successfully treated patients with VL rebounds have an increase in both their specific anti-HIV cellular and humoral responses compared to patients with maintained undetectable VL. 7 –9 These changes in immune responses could induce a better control of HIV replication after HAART interruption. If HIV-infected patients who receive multiple doses of vaccines present transitory increases in VL, it could be a good model to determine if such rebounds would increase specific responses against HIV, improving control of viral replication.
To test this hypothesis, 26 successfully treated HIV-infected adults were randomized to receive an immunization schedule with seven recommended vaccines or placebo. After the vaccination program, HAART was discontinued. The influence of vaccination over VL rebound, the risk of developing genotypic mutations, different T cell subsets, and HIV-1-specific immune responses were assessed during treatment and after it was discontinued.
Materials and Methods
Study design
A prospective, randomized, double-blind, placebo-controlled study was performed at the Hospital Clínic of Barcelona, Spain, from April 2003 to July 2006. Twenty-six HIV-infected patients successfully treated with HAART were randomized to receive either a vaccination program or placebo for 12 months. In month 12, HAART was interrupted in both groups. Treatment was reinitiated either if the CD4+ T cell count dropped below 350 cells/mm3 at any time after interruption or if VL increased above 5000 copies/ml after 6 months without HAART. Blood samples were taken monthly during treatment and after interruption. The primary end point was to determine if vaccinations increased the risk of viral rebound. Secondary end points were to determine if vaccinations increased the risk of developing resistance mutations, and if vaccinations induced HIV-1-specific immune responses improving control of HIV replication after HAART interruption. All patients provided written, informed consent. The present study was approved by the institutional ethics review boards and was registered in the public clinical trials database of the NIH with the number NCT00329251.
Study population
The eligibility criteria were as follows: age above 18 years, documented and asymptomatic HIV-1 infection, CD4+ T cell count above 500 cells/mm3 at least 6 months before inclusion, CD4 nadir above 300 cells/mm3, being under HAART at least 1 year before inclusion, VL below 200 copies/ml at least 6 months before inclusion, and VL previous to any treatment above 5000 copies/ml. Patients with basal creatinine above 2.5 mg/dl, transaminases above 250 IU/liter, chronic hepatitis B infection, allergy to either a whole vaccine or a component, and pregnant women were not included. Previous exposure to vaccination agents was registered but it did not influence the recruitment or randomization of patients. Participants were randomized into a vaccinated group (n = 13) or placebo group (n = 13) at month 0. The influence of vaccination on VL rebound, the risk of developing genotypic mutations, different cellular subsets, and HIV-1 specific immune responses were assessed during treatment and after it was discontinued by comparing these groups.
Immunizations
The vaccination program included 15 antigenic stimuli with 7 different usually recommended vaccines against 10 different agents (Fig. 1): hepatitis B (Engerix B, Smithkline Beecham SA; months 0, 1, 2, and 6), hepatitis A (Havrix 1440, Smithkline Beecham SA; months 4 and 10), influenza (2003–2004 WHO recommended vaccine [A/New Caledonia/20/99 (H1N1), A/Moscow/10/99 (H3N2), and B/Hong Kong/330/2001]; month 1), pneumococcal (Pneumo 23, Aventis Pasteur MSD SA; month 2), varicella (Varilrix, Smithkline Beecham SA; months 4 and 6), measles-mumps-rubella (Priorix, Smithkline Beecham SA; month 8), and tetanus-diphtheria (Ditanrix Adult, Smithkline Beecham SA; month 10). The placebo group received the same doses of placebo (0.5 ml of saline solution) at the same months.
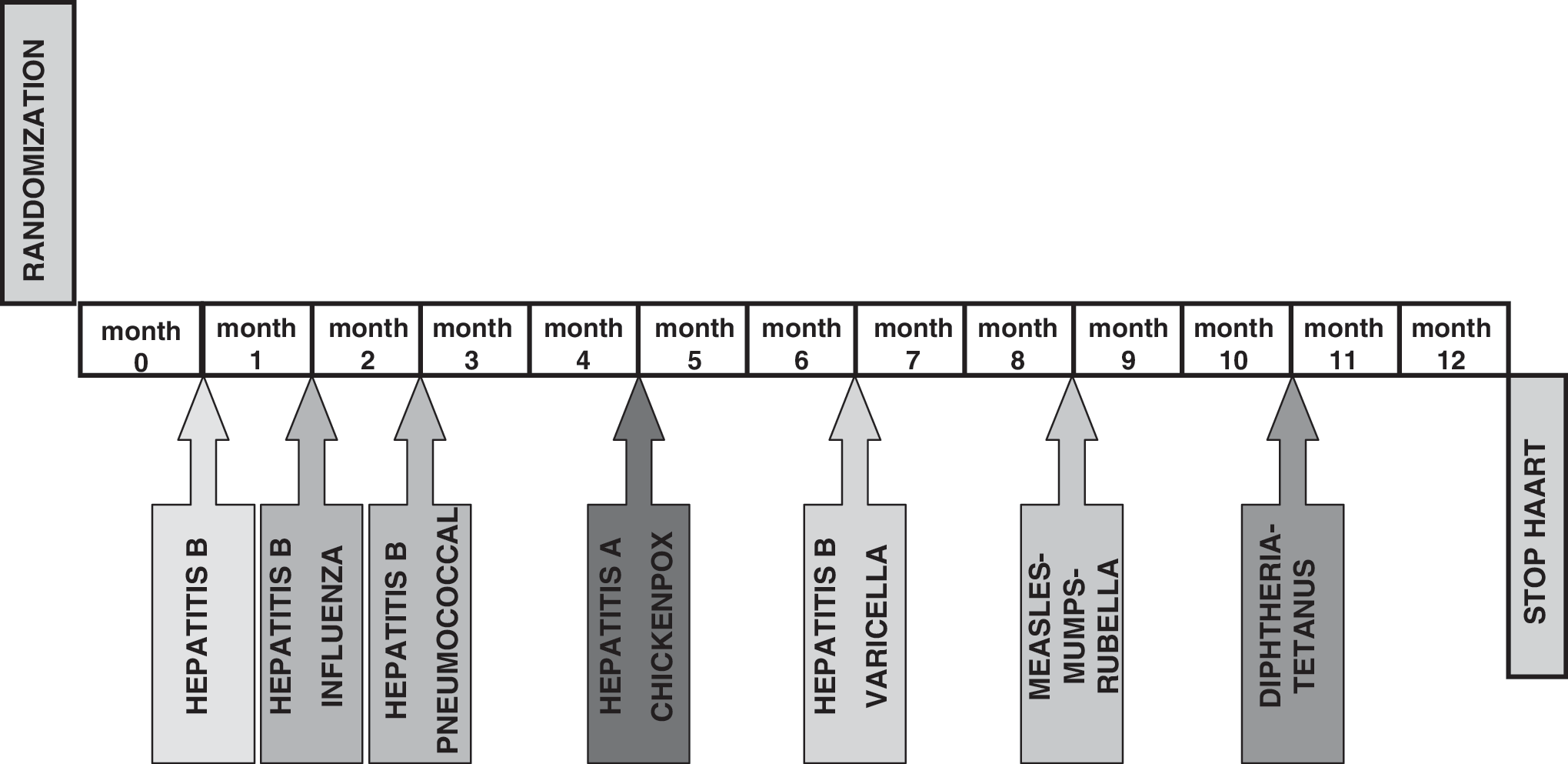
Vaccination program. It included 15 antigenic stimuli with 12 inoculations of 7 different usually recommended commercial vaccines against 10 different agents.
Virological evaluations
Plasma HIV-1 RNA levels were determined using the Amplicor HIV-1 Monitor Ultra Sensitive Specimen Preparation Protocol Ultra Direct Assay (Roche Molecular Systems, Inc., Somerville, NJ) with a limit of quantification of 200 copies/ml. Those samples below the detection limits of this test were retested with a lower limit of detection of 20 copies/ml. Population-based genotypic resistance testing was performed during treatment every time VL was over 1000 copies/ml and the first time VL was over 1000 copies/ml after HAART interruption, using the TruGene Assay (Visible Genetics).
Immunological evaluations
Different T cell subsets were determined as previously described using three-color flow cytometry. 10 Briefly, peripheral blood mononuclear cells (PBMCs) were obtained by separation on a Ficoll-Hypaque centrifugation gradient. Samples containing 105 cells were used for direct staining with the following monoclonal antibodies: CD3-PerCP, CD4-PerCP, CD8-PerCp, CD4-FITC, CD8-PE, CD28-PE, CD57-FITC, DR-FITC, CD38-PE, CD45RO-PE, CD45RA-FITC, CD62L-PE, CD25-PE, CXCR4-FITC, and CCR5-PE (Becton Dickinson, Mountain View, CA). The stained cells were analyzed on a FacSCalibur (Becton Dickinson, San Jose, CA) flow cytometer. Data were analyzed using CellQuest software.
PBMC proliferation assays (LPR) were performed essentially as previously described. 11,12 Briefly, cells were cultured in the absence or presence of phytohemagluttinin (PHA) (0.5 and 1%) 90 μg/ml (Murex, Biotech Ltd, England), OKT3 10 ng/ml (Ortho Biotech Inc., Raritan, NJ), anti-CD28 100 μg/ml, pokeweed mitogen 10 μg/ml (Sigma, St Louis, MO), tetanus toxoid 2750 U, cytomegalovirus (CMV) antigen 10 μg/ml, and 5 μg/ml of HIV-1 antigens gp160, p24 (Protein Sciences, Meriden, CT). Incorporation of tritium-labeled thymidine was assessed for the last 18 h of culture (Betaplate LKB Wallac, Sweden). Results were expressed as mean counts per minute (cpm). The stimulation index (SI) was calculated for each sample as cpm for cells with stimulus/cpm for cells without stimulus. A positive response to polyclonal stimulation was considered when SI was greater than 15. Positive antigen-specific responses were defined as more than 3000 cpm and SI greater than 3.
An ELISPOT (enzyme-linked immunospot) assay was used to measure HIV epitope-specific CD8+ T cell interferon-γ release from cryopreserved PBMC samples, as previously described. 13,14 Briefly, PBMCs were plated in the presence of different HLA class I-restricted synthetic peptides from gag, pol, env, and nef proteins. Spot-forming cells (SFC) were counted using an AID ELISPOT reader (Autoimmun Diagnostica GmHb, Germany). After subtracting background counts obtained with PBMCs and medium alone, results were normalized to SFC/106 PBMCs. A positive response was considered when counts were >40 SFC/106 PBMCs.
Statistical analysis
A total of 26 patients were recruited and a total of 15 antigenic stimuli inoculations were performed in each patient during 1 year (for a total of 195 stimuli). The sample size was calculated based on the total number of stimuli and assumed that the proportion of patients with VL rebound after one antigenic stimulus in the vaccinated group would be 20% and below 9% in the placebo group. 6 Assuming a type 1 error of 0.05 and a power of 80 two-sided nonparametric tests, this number will allow detection of a difference of 11% between the vaccinated arm and the placebo arm allowing for 20% of nonassessable patients.
Continuous variables are reported as medians and interquartile ranges (IQR). Comparisons between groups were made by using the Mann–Whitney U test for continuous data and the χ2 or Fisher's exact test for categorical data. Correlations were studied by using Spearman's rank correlation. Changes and durability in quantitative variables were analyzed by an area under the curve (AUC) measurement that incorporated the baseline value. Simple comparisons were made using a two-sided alpha level of 0.050; each of the pairwise comparisons used a two-sided significance level of 0.05/number of comparisons.
Results
Clinical characteristics
The median age of the included patients was 38.71 years (IQR 24.74–58.24). Among the 26 participants, 21 were male and 5 female, and the main risk factor for HIV infection was sexual (92%). There were no significant differences between the vaccinated and placebo group in clinical baseline characteristics (Table 1), although vaccinated patients were slightly younger than placebo patients (37.98 vs. 39.57; p = 0.064).
For quantitative variables median and interquartile range are expressed; for categorical variables, the number of patients and percentage.
Fisher's exact test.
Patients were under the following antiretroviral regimens (no differences between groups): stavudine + didanosine + nelfinavir (n = 1 patient), stavudine + didanosine + nevirapine (n = 2), lamivudine + tenofovir + efavirenz (n = 1), lamivudine + tenofovir + nevirapine (n = 1), lamivudine + stavudine + nelfinavir (n = 1), lamivudine + stavudine + nelfinavir, lamivudine + stavudine + lopinavir/ritonavir (n = 1), lamivudine + abacavir + nelfinavir (n = 4), lamivudine + zidovudine + abacavir (n = 1), lamivudine + zidovudine + efavirenz (n = 6), lamivudine + zidovudine + nevirapine (n = 1), lamivudine + tenofovir + lopinavir/ritonavir (n = 1), lamivudine + didanosine + nelfinavir (n = 6).
A vaccination and placebo schedule was maintained in all patients, except one who suffered accidental death unrelated to the protocol in month 9. There were neither local nor systemic secondary effects related to vaccination or placebo. One patient in the vaccinated group suffered acute hepatitis A infection in month 2. No clinical events appeared after HAART discontinuation.
Virological evolution
During the treatment period 314 VL determinations were performed, and 22 of them were detectable above 200 copies/ml. The incidence was 0.07 detectable VL/determination/year, 14 in the placebo group (incidence 0.091; 95% CI 0.051–0.15) and 8 in the vaccinated group (0.05; 95% CI 0.022–0.095). No significant differences between groups were found [p = 0.22, risk ratio (RR) of 0.54 (95% CI 0.23–1.26)]. Detectable determinations were observed in five patients in each group. In the vaccinated group, three detectable VL rebounds were preceded by a vaccination, but five were not. The range of magnitude of detectable VL was between 210 and 51,200 copies/ml, median 544.5 copies/ml. By groups, range and median were 237–15,900 and 584.5 and 210–51,200 and 471 copies/ml in the placebo and vaccinated groups, respectively. The influence of the result of previous serology against the inoculated vaccine in VL rebound was also analyzed. Patients without previous contact with the agent did not have a higher risk of VL rebound after vaccination than patients with a previous positive result (data not shown).
When samples with VL below 200 copies/ml were reanalyzed with an assay with a limit of detection of 20 copies/ml, the total number of detectable VL increased to 49 determinations (incidence of 0.156, 95% CI 0.12–0.2), 31 in the placebo group (0.20, 95% CI 0.14–0.27) and 18 in the vaccinated group (0.11, 95% CI 0.068–0.17) [p = 0.04, RR 0.55 (95% CI 0.32–0.94)], distributed in seven and six patients, respectively (with a mean of 4.42 detectable VL/detectable patient in the placebo group and 2.86 detectable VL/detectable patient in the vaccinated-group, p = 0.249) (Fig. 2A). In the vaccinated group, 7 detectable VL determinations were preceded by a vaccination in the previous month, but 11 were not.
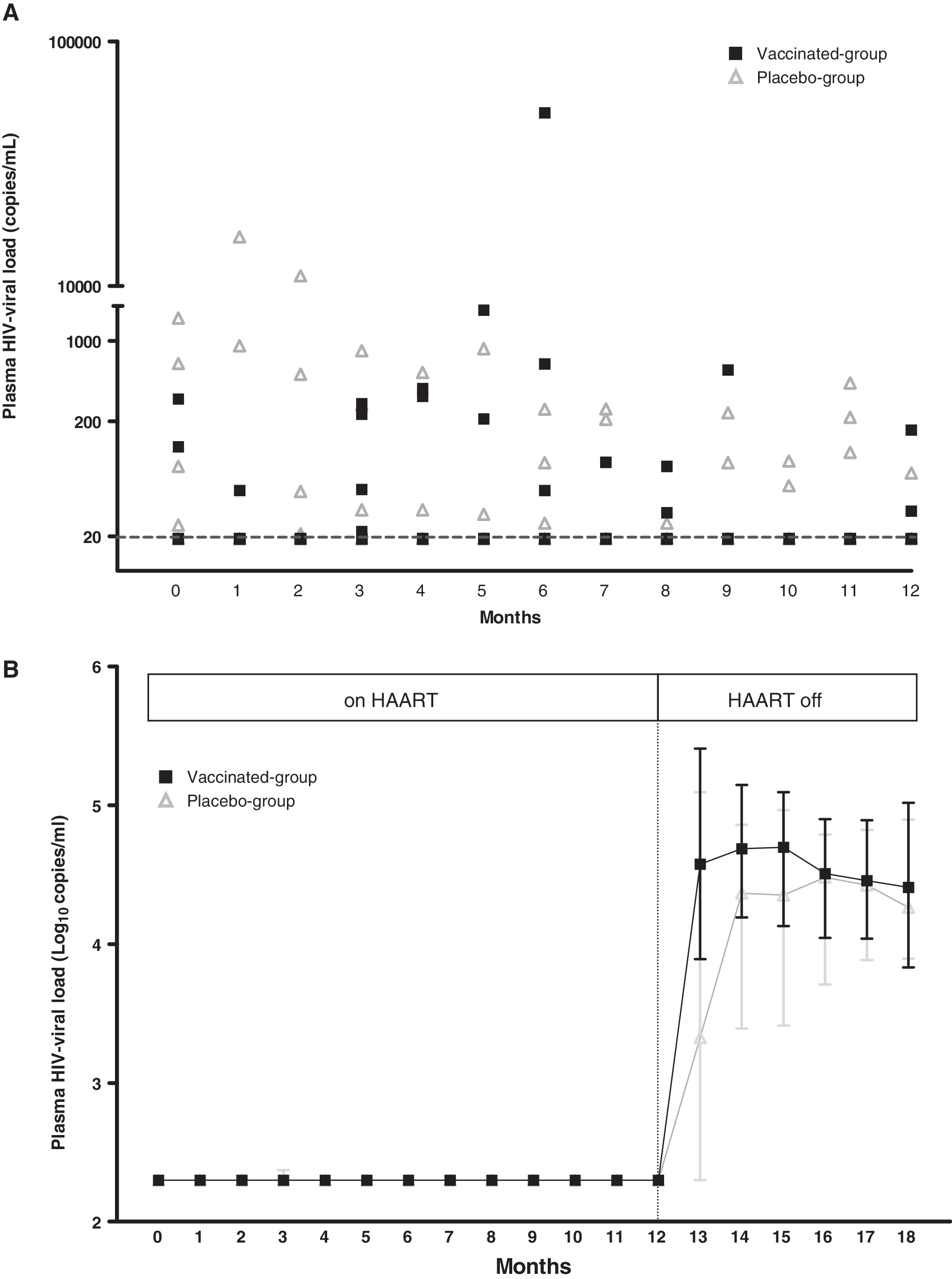
(
Two patients changed HAART during the study period. One patient in the placebo group receiving lamivudine, abacavir, and nelfinavir presented persistent low level viremia in five consecutive determinations (range 87–856 copies/ml). It was not possible to isolate the virus for genotyping; treatment was changed to efavirenz, didanosine, and tenofovir. In addition, another patient from the vaccinated group receiving lamivudine, didanosine, and nelfinavir presented a progressive increase in VL since 51,200 copies/ml was associated with an adherence of 75%. Virus genotyping showed an M184V mutation. In this case, HAART was changed to tenofovir, didanosine, and nelfinavir and VL returned to an undetectable level after 1 month.
After HAART discontinuation, a rebound of VL was observed in all the patients. No significant differences between groups were observed in VL at months 13 or 18 (p = 0.18, 0.94, respectively), peak VL (p = 0.83), and the AUC of the VL rebound between both groups (p = 0.3) (Fig. 2B). A study of genotype was performed in 22 patients (11 in the vaccinated group and 11 in the placebo group). No clinically relevant mutations in the retrotranscriptase or protease genes were detected.
Changes in T cell subsets
At baseline, vaccinated patients had a higher proportion of CD4+CD38+ and CD4+CD28+ and a lower proportion of CD4+CD45RA−CD45RO+ T cells. No differences in other T cell subsets were observed (Table 2).
Median and interquartile range are expressed.
Measured by stimulation index.
Measured by spots forming cells/106 peripheral blood mononuclear cells (SFC/106 PBMCs).
During the study period on HAART, while CD4+ T cells increased a median (IQR) of 5.46% (29.11) in the placebo group, it decreased 12% (21.49) in the vaccinated group (p = 0.034, for the difference between groups) (Fig. 3A). Apart from that, activation markers as measured by CD4+CD38+, CD8+CD38+, and CD8+CD38+HLADR+ increased significantly in the vaccinated group (p = 0.028, 0.002, and 0.001, respectively, for the difference between month 12 and baseline value) and not in the placebo group. It was noticed that changes of these subsets tended to be different between groups (p = 0.07, 0.06 and 0.09) (Fig. 3B–D). The decrease of CD4+ T cells was correlated with a CD4+CD38+ T cells increase (p = 0.044, r = −0.397). Finally, the CD8+CD45RA+CD45RO− population decreased in vaccinated patients and increased in placebo recipients (p = 0.02, for the difference between groups). The rest of the T cell subpopulations evolution did not differ significantly between groups (Table 2).

Evolution of immunological parameters during the study period. Months 0 to 12, patients were on HAART. At month 12 HAART was discontinued (shady area) [▪ vaccination group, Δ placebo group]. (
After HAART was interrupted, only one patient in the vaccinated group had to restart therapy in month 18 because the CD4+ T cell count fell below 350 cells/mm3.
Different T cell subsets had different evolutions in the vaccinated group and the placebo group in the period after HAART interruption. These differences were observed mainly in the first 2 months after interruption (months 12 to 14). The decrease in CD4+ T cells and the increase in the CD4+CD38+HLADR+ population were lower and later in the vaccinated group than in the placebo group (p = 0.015 and 0.035, respectively). The increases in CD8+ T cells (p = 0.034), CD8+CD38+ T cells (p = 0.003), and CD8+CD38+HLADR+ T cells (p = 0.033) were higher and sooner in the vaccinated group than in the placebo group. Naive (CD45RA+CD45RO−) and memory (CD45RA−CD45RO+) T cells also differ in their evolution in these months: the CD8+CD45RA+CD45RO− subset decreased more in the placebo group than in the vaccinated group (p = 0.039), and CD8+CD45RA−CD45RO+ increased more in the vaccinated patients (p < 0.05) during the first 2 months of interruption. All these differences tended to disappear at month 18 (6 months after the interruption) (Table 2 and Fig. 3A–D).
Lymphoproliferative responses evolution
No baseline differences in LPR to polyclonal mitogens, recall, and HIV antigens were found between groups. In the period of treatment, LPR to PHA and p24 HIV antigen increased in the vaccinated group compared to placebo (p = 0.009 and p = 0.04, differences in the AUC between groups). Changes in LPR to recall antigens or gp160 HIV antigen were not observed.
After interruption of HAART, a lower and later increase in LPR to PHA (p = 0.027) and p24 HIV antigen (p = 0.05) was observed in vaccinated patients compared to the placebo group (Table 2 and Fig. 3E–F).
HIV-specific CD8+ T cell responses evolution
The vaccinated and placebo group showed similar HIVspecific CD8+ T cell responses at month 0 and no significant differences during the treatment period were observed.
After HAART interruption both the magnitude and the breadth of the total HIV-specific CD8+ T cell responses showed a trend to increase more in the vaccinated group, mainly against the p17 and gag peptides pool (p = 0.043 and p = 0.063, respectively) (Table 2 and Fig. 3G–H).
Discussion
Exposure to 15 antigenic stimuli against 10 different agents (for a total of 195 stimuli) was not associated with an increase in VL rebound in our study, neither the whole schedule nor an individual vaccine, irrespective of baseline serological status. Vaccinations were not associated with clinical adverse events and were not associated with the development of genotypic resistance mutations. Conversely, the vaccinations were accompanied by changes in some immunological parameters that did not influence VL rebound dynamics after HAART interruption.
As HIV-infected patients are considered immunosuppressed individuals independently of CD4+ T cell count, they are advised to be vaccinated. Current recommendations propose combining several vaccinations in one visit in order to reduce the number of interventions and to not lose the opportunity of vaccinate a patient. 15 Sometimes a patient's previous serological status is not known and it is not tested (i.e., vaccination against hepatitis B and tetanus in an emergency room after parenteral exposure to supposedly contaminated material). 16 The schedule used in this study was safe and was not associated with local or systemic effects, although some patients have had previous exposure to some of the vaccines.
Vaccination did not influence the incidence of detectable VL. Although it has previously been indicated that vaccines are associated with increases in VL 1,2,17 and development of resistance to HAART, 6 some controversy exists. We did not find differences between groups, in spite of the increase in activated T cell subsets. Even, when samples with VL below 200 copies/ml were reanalyzed with an assay with a limit of detection of 20 copies/ml, we found a higher number of detectable VL in the placebo group, which was due to a higher number of detectable VL episodes in the “detectable patients” in this group. Randomization equilibrated variables that could have influenced the presence of transient increases of VL, such as the time of infection, time under HAART, number of previous lines of treatment, previous use of monotherapy or biotherapy, and nadir CD4+ T cells. 18 –25
Increases in VL shorter than 30 days of duration 26 or limited to the lymphatic compartment would not have been detected. However, if they had appeared, they did not seem to have clinical significance because VL returned to an undetectable level 1 month after vaccination and they did not increase the risk of developing genotypic mutations. Therefore, it is likely that a vaccination schedule does not influence clinical progression in successfully treated HIV patients.
In the present study, vaccines induced changes in the immunological system, with the main finding being the decrease in CD4+ T cells. This fact has been previously described in the pre-HAART era and with a unique vaccination. 27,28 This decrease seems to be associated with an increase in CD4+ T cell activation more than with an increase in viral replication, as suggested by the correlation between the decrease of CD4+ T cells and the increase of CD4+CD38+ T cells. Activation is considered a major determinant in CD4+ T cell depletion in HIV infection, 29 –32 and this study could be considered an in vivo model that supports this information.
An increase of the LPR was also observed in the vaccinated group. As this increase presented with polyclonal mitogens as well as with HIV proteins, it seems to suggest a polyclonal T cell activation due to vaccines. Recent publications have found that exposure to one antigen can activate both specific and unspecific T cells. 33 In addition, we did not find an increase in HIV-specific CD8+ T cell responses during the treatment period associated with the vaccination schedule. These data could be a potential explanation of the lack of VL control after HAART interruption despite the increase in anti-HIV-specific LPR in the vaccinated group. There are contradictory data about the influence of anti-HIV-specific LPR in VL rebound after HAART interruption. Although some authors have found that the increase of these responses is associated with a better control of VL after HAART interruption, 34 –36 other reports have indicated that a high HIV-1-specific LPR did not influence the clinical outcome of the patients. 37 –39 Explanations for that paradox have been the existence of parallel dysfunctions in natural killer immune responses, 40,41 clonal depletion, 42 or impaired CD8+ T cell function. 43
After HAART interruption, although the changes in immune systems observed were similar to those previously described after interrupting HAART, 10,44,45 vaccinated patients showed a different immunological evolution in the early phase of interruption (months 12 to 14), although these changes tend to disappear at the end of the follow-up period (month 18). A lower and later CD4+ T cell activation, a decrease in CD4+ T cells, and an increase in LPR were observed in vaccinated patients. A higher and quicker increase in CD8+ T cell activation, CD8+ T cells, and HIV-specific CD8+ T cell responses was also shown. Although these differences were short term, they reflect an effect of vaccines on the immune system of HIV patients.
Interestingly, when both time periods (“on HAART” and “off HAART”) are analyzed in the two groups, T cell subset evolution in the vaccinated group during the period on HAART and after HAART interruption looked very similar to those in the placebo group after HAART interruption. This could suggest that those changes in T cell subsets are caused by antigenic stimulation, which appears in different times in both groups because of its different origin: in the placebo group by the viral load rebound after HAART interruption and in the vaccinated group by the vaccinations during the period on HAART and the viral rebound after stopping HAART.
It has to be pointed out that although one of the objectives of the study was to evaluate the effects of vaccination after HAART interruption, to determine if it was possible to have a large interruption in HAART, recent evidence suggests that drug interruptions are no longer appropriate and should not be undertaken. 46
Although the design of the study, prospective, randomized, and double blind controlled with placebo (which is used only in a few studies that explore the influence of vaccinations in VL 47 –52 ), gives power to our results, this study has several limitations. First, one important limitation is the size sample. Although it was calculated in order to describe the influence of vaccination over VL rebound, 6 it could be too small to determine differences in other parameters as variations in T cell subsets, LPR, HIV-specific CD8+ T cell responses, or the magnitude of the rebound in VL after discontinuing HAART. It has also to be noted that although in the case of VL, vaccinated patients received 195 antigenic stimuli more than the placebo group, it is possible that observations within individuals are correlated and it could influence the results. Second, because of the absence of stratification during randomization, vaccinated patients tended to be younger. This could explain why some immunological parameters at baseline differed between the vaccinated and placebo group, as naive T cells, which are known to be higher in younger people. Third, patients included in our study were in a “preserved” immune status because of inclusion criteria. Vaccinations in patients without treatment or more immunosuppressed could suppose dramatic changes in the results, and therefore they cannot be generalized to that population. Finally, as we have administered a vaccination schedule, we could have analyzed the effect of this scheme and not the effect of each different vaccine in particular. Although it is possible that each vaccine could have different effects on the immune system, as suggested by some changes observed in the immune responses depending on the previous vaccination, the design of the study does not allow for discriminating it.
In conclusion, a vaccination schedule in successfully treated HIV-infected patients would be safe, and would not be associated with clinically relevant VL rebound or the development of genotypic mutations. However, it would induce changes in the immune system with T cell activation and a decrease in CD4+ T cells, as well as an increase in anti-HIV immune responses, although these changes would not be associated with a change in the dynamics of VL rebound after HAART interruption.
Footnotes
Acknowledgments
We would like to thank Dr. Jordi Yagüe (Immunology Department) for help with the determination of humoral responses and all the laboratory technicians involved in the project and nursery for help with the Adult Vaccination Center. Finally, we specially thank the study subjects for their time and participation in this study. This study was supported in part by Grants ISCIII-RETIC RD06/006 [Red Temática Cooperativa de Grupos de Investigación en SIDA del Fondo de Investigación Sanitaria (FIS)], FIPSE [FIPSE is a nonprofit foundation including the Spanish Ministry of Health, Abbott Laboratories, Boehringer Ingelheim, Bristol Myers Squibb, GlaxoSmithKline, Merck Sharp and Dohme, and Roche] 36536/05, SAF 05/05566, FIS PI050058, FIT 090100-2005-9, FIS PI050265, and FIS 04/0503. Dr. Felipe García was a recipient of a research grant from IDIBAPS, Barcelona, Spain. Dr. Montserrat Plana was supported by contract FIS 03/00072 from the Fundació Privada Clínic per la Recerca Biomèdica in collaboration with the Spanish Health Department. Part of the information contained in this manuscript has previously been presented at the 12th Conference on Retroviruses and Opportunistic Infections (February 2005, Boston, Massachusetts), abstract #523.
Disclosure Statement
No competing financial interests exist.