
Abstract
We have identified a subset of HIV-susceptible CD4+CCR5+ cells in human PBMCs that can efficiently exclude protease inhibitors (PI) due to high P-glycoprotein (P-gp) efflux activity. Phenotypically these cells are heterogeneous, include both T and non-T cells, and some display markers of memory cells. Cells with high P-gp represent 16–56% (median = 37.3) of all CD4+CCR5+ cells in healthy donors, and are selectively depleted in HIV-1-infected individuals (4.1–33%, median = 10.1). A fraction of primary cells productively infected by HIV-1, in vitro, have high P-gp pump activity, demonstrating that infection does not inhibit P-gp function. In agreement with these data, HIV-susceptible cells expressing high levels of P-gp require higher levels of PI for complete inhibition of virus spread. We conclude that the PI concentrations achieved in plasma could be suboptimal for full inhibition of virus spread in high P-gp cells, indicating that they may represent a pharmacological sanctuary for HIV-1.
Introduction
T
It has been shown that highly active antiretroviral therapy (HAART) does not completely inhibit viral replication 15 ; low levels of viremia have been detected by the use of more sensitive polymerase chain reaction (PCR) protocols 16 –18 and by in situ hybridization in samples from lymphatic tissue in patients under HAART. 19 One possible contributor to the chronic low levels of HIV-1 replication may be the presence of cells in which the antiviral effects of the drugs are suboptimal. Cells within the brain and genitourinary tract have been studied as anatomical sanctuaries for HIV-1, due to the blood–brain and blood–testes barrier. 20 –26
Drug efflux pumps, such as P-glycoprotein (P-gp), are important for drug resistance development. P-gp is a plasma membrane protein, encoded by the multidrug resistance type 1 (MDR-1) gene, 27 with the ability of transporting a wide variety of lypophilic compounds out of the cell. P-gp function is ATP dependent, and its activity decreases the concentration of transported drugs at intracellular target sites, resulting in therapeutic failure and multidrug resistance. P-gp activity is high in several anatomical sites such as liver, pancreas, gut, kidney, and the blood–brain barrier. 28 At the single cell level, P-gp activity is heterogeneous among cells of the immune system, with high levels in cells with cytolytic function, i.e., natural killer (NK) cells and cytotoxic T-lymphocytes. 29 –32 Because PIs are substrates for P-gp, 33 –36 the pharmacokinetics and intracellular concentration of these drugs are influenced by P-gp activity in different cellular subsets and anatomical sites. 37 We tested the hypothesis that high P-gp activity in a subset of HIV-1 target cells results in PI exclusion, thus potentially contributing to treatment failure. We identified a subset of CD4+CCR5+ lymphocytes with high P-gp activity, and found that these cells are selectively depleted in HIV-1-infected individuals. PIs showed suboptimal inhibition of HIV-1 replication in infected primary cells with high levels of P-gp. Therefore, the presence of such cells in HIV-1-infected individuals receiving HAART may lead to suboptimal intracellular drug levels. Continuous low-level virus replication in such cells may eventually lead to drug resistance and ultimate treatment failure.
Materials and Methods
Clinical samples and cell isolation
Blood samples were collected in ACD tubes under approved protocols for human subjects' research by the National Cancer Institute Investigational Review Board. Most patients included in this study were under HAART, usually including at least one PI and had viral loads below the detection of the assay. Peripheral blood mononuclear cells (PBMCs) were obtained by gradient centrifugation over Histopaque (Sigma) gradients according to the manufacturer's protocol.
Flow cytometric analysis
PBMCs were washed twice with phosphate-buffered saline (PBS) containing 0.5% heat-inactivated human AB serum (Sigma). The cells were incubated for 20 min at 4°C in the dark with directly conjugated monoclonal antibodies (Pharmingen). Cells were washed twice and analyzed in an LSR flow cytometer. The data were analyzed using Cell-Quest software. In this study, we used a panel of directly conjugated antibodies (BD-Pharmingen) recognizing the following human antigens: CD3, CD4, CD8, CD14, CD19, CD56, CCR5, and CD45RO.
Viral infection
The green fluorescent protein (GFP)-tagged HIV-1 molecular clones NL4-3GFP and JR-CSFGFP were used for in vitro infection experiments. Infectious stocks were generated by transfection in 293 cells as described. 38 Infected cells were cultured in RPMI 1649, supplemented with 10% fetal bovine serum (FBS) and 5 units/ml of human recombinant interleukin-2 (IL-2) at a density of 106 cells/ml. Viral replication was monitored by measuring HIV-1 p24gag accumulation in culture supernatants using a commercial ELISA kit (Zeptometrix) and by inspecting the frequency of GFP+ cells by flow cytometry.
P-gp pump activity
Rhodamine123 (Rh123) and tetramethylrhodamine methyl ester (TMRE), two fluorescent rhodamine derivatives that are substrates for the P-gp pump, 39 were used in flow cytometric studies of the P-glycoprotein activity in primary lymphocytes. This method allows functional studies of pump activity combined with the immunophenotyping of different cell subsets in a heterogeneous population such as PBMCs. Briefly, for loading of the dye, PBMCs were resuspended at a density of 107 cells/ml in RPMI medium supplemented with Rh123 (0.5 μg/ml) and incubated at 37°C for 20 min. After washing to remove the nonincorporated dye, the cells were resuspended in medium without Rh123 and kept at 37°C to allow the cells to pump out the dye. At different time points aliquots were taken, stained with conjugated antibodies, and analyzed by flow cytometry. CsA, Verapamil, and Cimetidine (Sigma) were used in inhibition experiments at concentrations known to specifically and completely inhibit ABC transporters (1 μM, 10 μM, and 50 μM, respectively).
Cell sorting
PBMCs were loaded with Rh123 and after 1 h of incubation at 37°C in RPMI in the absence of the dye, the cells were stained with directly conjugated antibodies against CD3 and CD4 (BD-Pharmingen). CD4+ T cells were sorted on a FACSAria flow cytometer based on their high or low Rh123 content. The purity of the sorted populations was verified using FlowJo Software (Treestar, Ashland, OR).
Real-time quantitative PCR
To determine whether P-gp activity directly correlates with the MDR1 levels of expression, total RNA was purified from sorted CD4+ T lymphocytes with different levels of functional P-gp using RNA-Bee (Tel-test, Inc., Friendswood, TX) according to the manufacturers's protocol. Reverse transcription was performed with a Superscript III reverse transcription kit (Invitrogen, Carlsbad, CA) using oligo(dT) primers. Quantitative PCR was performed using commercially available kits specific for human MDR1 and GAPDH containing premixed primers and specific probes (Invitrogen). Amplification was performed for 40 cycles on an ABI Prism 7700 (15 s at 95°C followed by 1 min at 60°C).
Results
High frequency of P-gp expression in CD4+CCR5+ primary cells
To identify cells capable of excluding anti-HIV drugs, we studied the activity of the P-gp efflux pump in human PBMCs, especially in primary CD4+ cells. Cells were loaded with Rh123, a fluorescent dye transported out of the cell by P-gp, and stained with several conjugated antibodies prior to flow cytometric analysis. P-gp activity was heterogeneous among primary cell subsets: NK cells and CD8+ lymphocytes were characterized by high efflux activity. In spite of the low P-gp activity present in the total CD4+ cell population, a subset of CD4+ cells with high P-gp activity was identified in both healthy controls (Fig. 1A) and HIV-1-infected individuals (see Fig. 2). Control experiments using verapamil, a known P-gp inhibitor, or cyclosporin A (CsA) efficiently blocked Rh123 efflux, whereas cimetidine, an inhibitor of the organic cation carrier, failed to inhibit Rh123 efflux (data not shown).
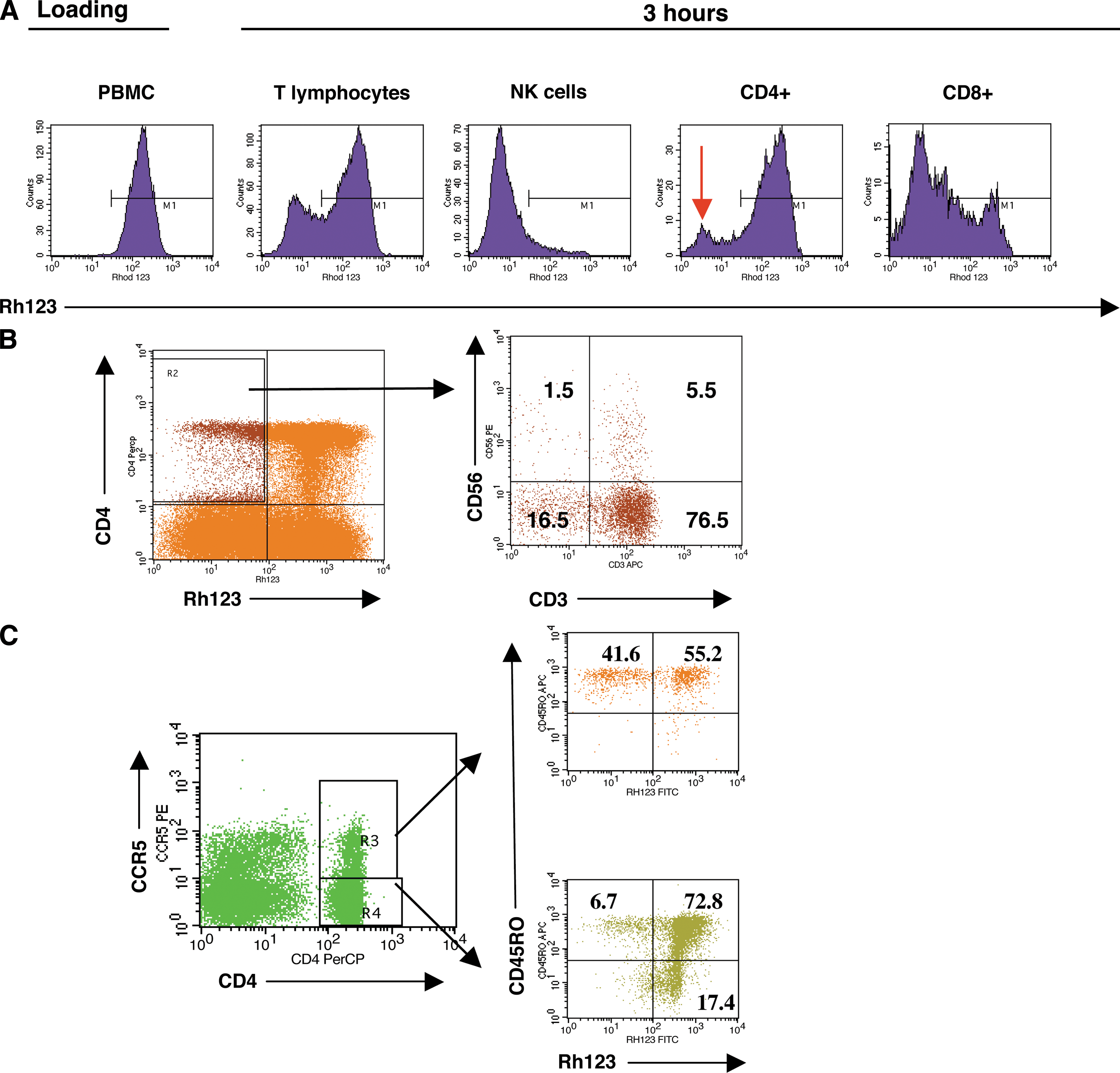
Analysis of P-gp efflux pump in human primary PBMCs. (
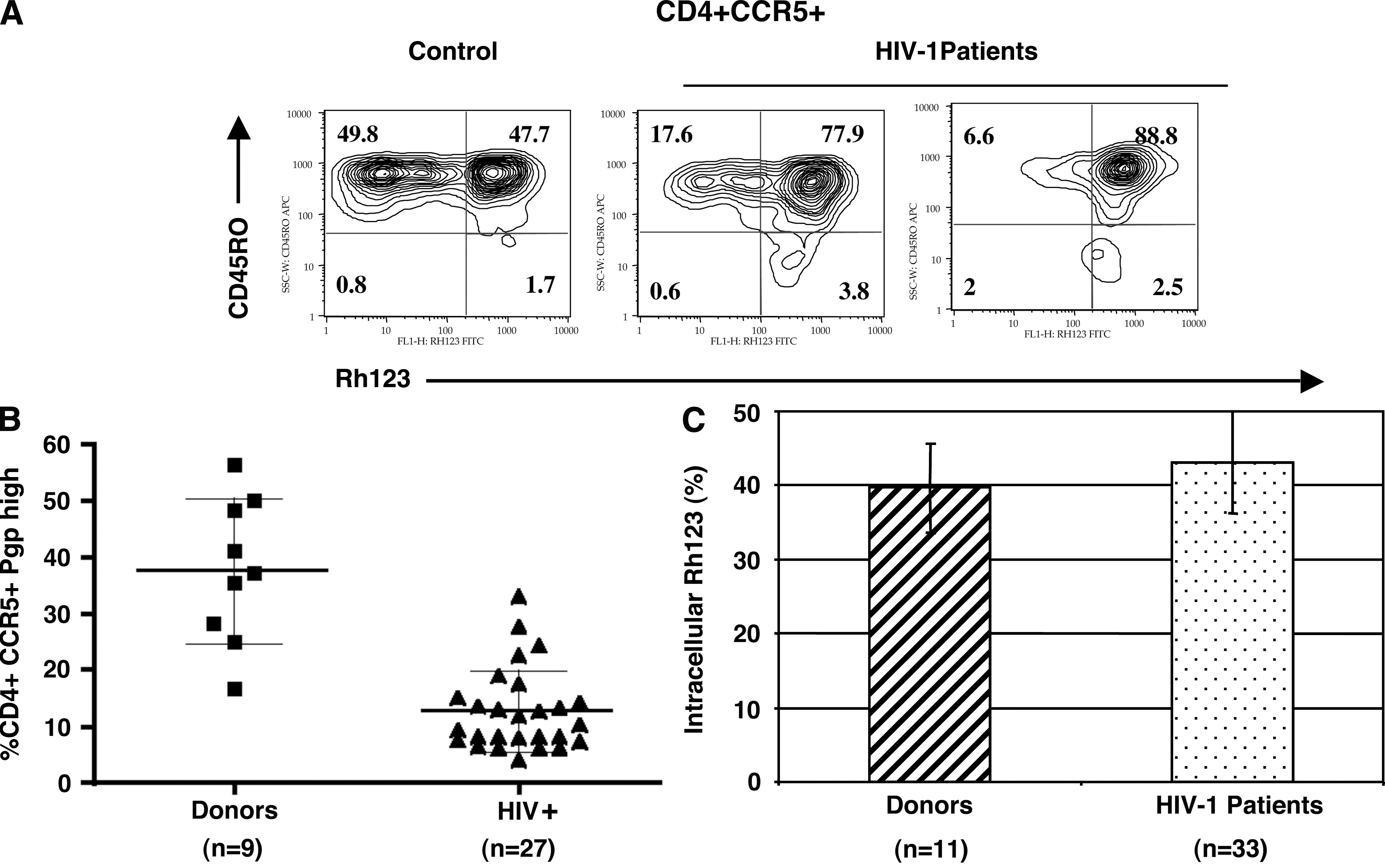
P-gp activity in primary CD4+CCR5+ cells and PBMCs of healthy donors and HIV-infected individuals. PBMCs were loaded with Rh123 and the amount of intracellular dye was measured by flow cytometry after 3 h of incubation in Rh123-free medium. Cells were stained with anti-CD4, anti-CCR5, and anti-CD45RO antibodies prior to flow cytometric analysis. (
To study whether there was a correlation between P-gp activity and MDR-1 mRNA, CD4+ cells were sorted according to high or low Rh123 content after 3 h in the absence of dye. The two populations were analyzed for the amount of MDR-1 mRNA normalized for GAPDH mRNA. We found that the cells rapidly excluding the Rh123 dye (Rh123 low) had 10- to 20-fold higher MDR-1 mRNA levels compared to the Rh123 high population, indicating a correlation between mRNA and functional protein levels of P-gp. Immunophenotyping of the CD4+P-gp(high) population using CD3 and CD56 surface staining 40 showed that it is heterogeneous, and contains T (both CD3+CD56− and CD3+CD56+ cells) and NK (CD3−CD56+) cells (Fig. 1B). Additional analysis of CD4+ T cells according to their CCR5 expression demonstrated that the CD4+P-gp high cells are mainly in the CCR5+ population (Fig. 1C), which makes them potential targets for HIV-1 infection. Interestingly, most of the CD4+CCR5+ cells with high P-gp activity were identified as memory T cells based on their expression of CD45RO (Fig. 1C).
CD4+CCR5+ cells with high P-gp levels are selectively depleted in HIV-1+ individuals
We measured the frequency of CD4+CCR5+ cells expressing high P-gp activity in healthy donors and HIV-infected individuals under combination drug treatment. Representative examples for this analysis including one donor and two infected individuals are shown in Fig. 2A. CD4+CCR5+ T cells expressing high P-gp represented 16–56% of the CD4+CCR5+ population in healthy controls (median = 37.3, n = 9) (Fig. 2B). The frequency of the CD4+CCR5+P-gphigh cells was selectively reduced in patients (4.1–33%, median = 10.1, n = 27), and this selective depletion was found in both patients receiving PIs (n = 20) and patients without PIs in their drug regimens (n = 7). The difference in healthy donors versus HIV-1-infected patients is significant (p = 0.0004, unpaired t test not assuming equal variances), indicating that these cells may be more susceptible to the deleterious effects of HIV-1 infection compared to the subpopulation expressing the same markers but no P-gp.
P-gp function is conserved in the total PBMCs of HIV-1-infected individuals
Several reports have indicated that P-gp activity is decreased in PBMCs from HIV-1-infected persons, 41 –43 which suggests that cells with high P-gp pump may be few and might not present a problem for drug activity against HIV-1. We therefore examined the function of the P-gp pump in a population of HIV-1-infected persons under therapy with drug combinations including PIs (n = 33), and compared it to healthy uninfected individuals (n = 11). We found that PBMCs from HIV-1-infected individuals incorporate more Rh123 on loading (20% more dye on average), probably as a result of cell activation leading to a higher accumulation of dye in the mitochondria. The percentage of Rh123 remaining intracellular 3 h after loading was similar in patients and controls (Fig. 2C), demonstrating that P-gp function was conserved in cells from HIV-1-infected individuals. Therefore, in spite of higher dye uptake, P-gp expression appears to be unaffected in the population of total PBMCs, based on the percent drug efflux in our experiments. These results show that there is no global impairment of P-gp activity in HIV-1-infected individuals. Rather, there is a selective depletion of cells expressing P-gp among cells that are targets for HIV-1 infection.
CD4+P-gphigh T cells actively replicate HIV-1
The presence of CD4+CCR5+ cells with high P-gp activity in HIV-1 patients does not exclude the possibility that productive HIV-1 infection results in impaired P-gp-mediated efflux. To study whether cells productively infected with HIV-1 display high levels of P-gp activity, we infected primary PBMCs with HIV-1 molecular clones tagged with GFP. Infected cells accumulate GFP as a result of viral expression and can be easily identified by flow cytometry. 38 To follow P-gp activity in infected cells simultaneously with GFP, we used the rhodamine derivative TMRE, a P-gp substrate with emission spectrum shifted towards the red 39 (Fig. 3). Infection of primary PBMCs with GFP-tagged HIV-1 molecular clones typically resulted in 1–2% of the cells expressing GFP as result of HIV-1 expression. We found that 15–33% of productively HIV-1-infected cells (GFP+) in different samples express high P-gp as demonstrated by TMRE efflux. The frequency of infected cells with high P-gp activity is in agreement with that of the CD4+CCR5+ population discussed above. These cells are heterogeneous and were identified as NK cells and T-lymphocytes based on their CD3 and CD56 surface staining. A minority of infected cells with high P-gp activity was negative for both T cell and NK cell markers. The nature of these cells remains to be determined.
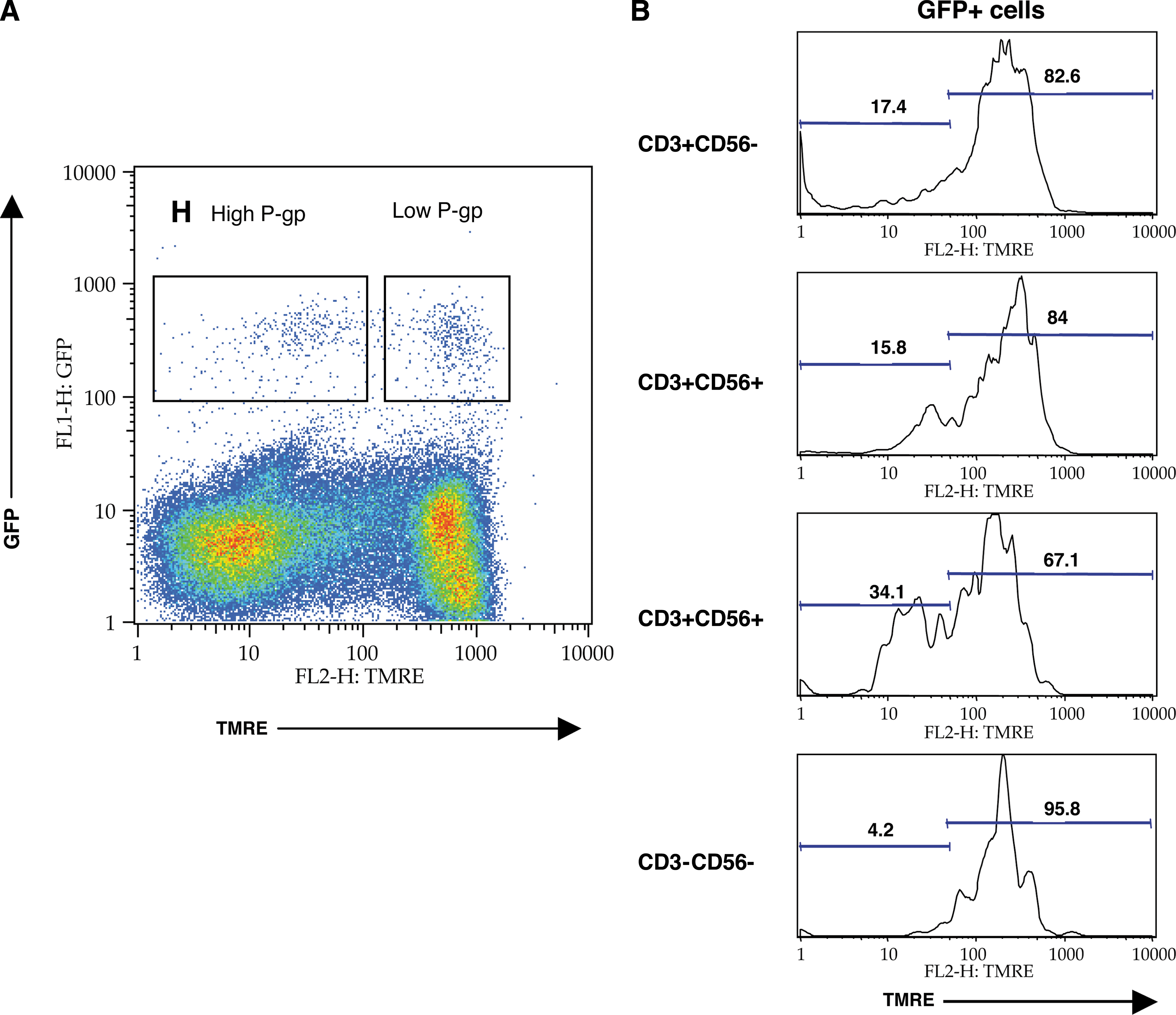
Identification of a subset of HIV-1-infected PBMCs with high P-gp pump activity. (
Decreased antiviral activity of PIs in CD4+ primary cells with high P-gp activity
To determine whether high P-gp activity affects the antiviral effects of PIs, primary CD4+ lymphocytes from a healthy donor were sorted based on their Rh123 content 3 h after loading and washing (Fig. 4A). The two sorted cell populations (P-gp high and low) were infected with HIV-1 in vitro (moi of 0.1) and cultured in the presence of different concentrations (5 and 50 nM) of the PI ritonavir. Infected cells were kept in culture for 12 days and viral replication was monitored by measuring p24gag accumulation in culture supernatants. Partial inhibition of HIV-1 propagation was found in the presence of 5 nM ritonavir in CD4+ cells with low P-gp activity and complete viral block was seen at 50 nM. In contrast, in cells with high P-gp activity, 50 nM ritonavir provided only partial protection, whereas 5 nM ritonavir failed to provide any antiviral effects on these cells (Fig. 4B). These results demonstrate that P-gp expression decreased the susceptibility of CD4+ primary cells to the antiviral effects of PIs.
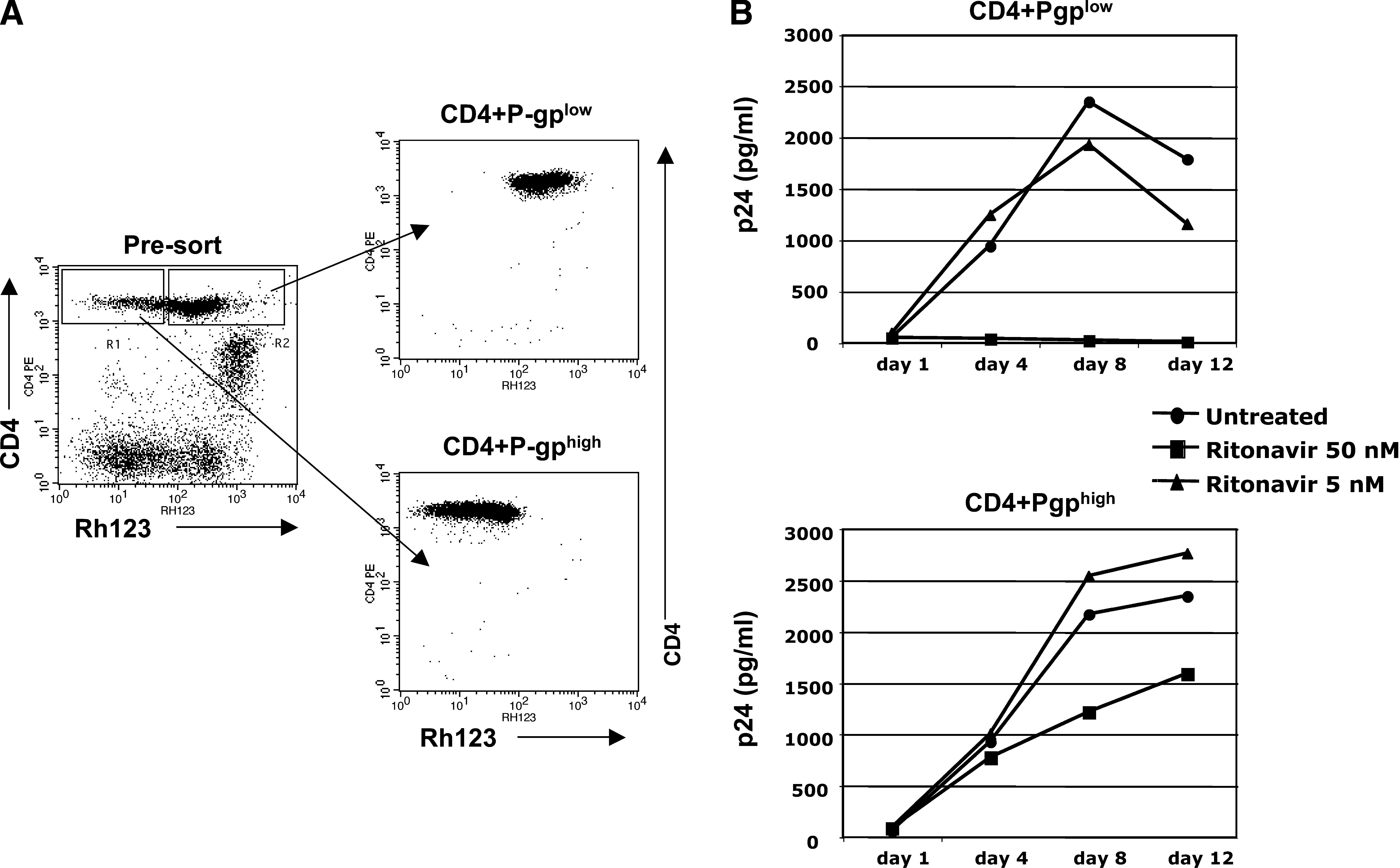
Suboptimal antiviral effects of ritonavir in primary lymphocytes with high P-gp activity. Primary CD4+ T cells from a healthy donor were sorted according to high or low P-gp activity, as judged by their Rh123 intracellular content after 3 h of incubation in medium without the dye. (
Protease inhibitors inhibit P-gp activity in a dose-dependent manner
P-gp substrates are also inhibitors of P-gp activity to various degrees. To study the effects of PIs on P-gp function, we monitored the efflux of Rh123 in the continuous presence of increasing PI concentrations from 5 to 135 μM (approx. 4–100 μg/ml), which exceed the plasma levels found in patients. Oral administration of PIs results in peak plasma levels of 1–23 μg/ml and trough plasma levels of <1–16 μg/ml. 46 –50 Both indinavir and ritonavir inhibited P-gp-mediated efflux of Rh123 in a dose-dependent manner (Fig. 5A and B), although ritonavir was found to be a more potent P-gp inhibitor than indinavir. Comparison of Rh123 efflux in NK cells and T-lymphocytes from the same PBMC sample indicated that similar levels of inhibition require higher drug concentrations in NK cells. Figure 5C shows that the P-gp inhibition curve by both PIs (indinavir and ritonavir) is shifted to the right in NK cells, demonstrating that higher concentrations are needed in NK cells to obtain similar biological effects. Ritonavir concentrations in the range found in plasma resulted in significant P-gp inhibition in PBMC samples. Despite this, a subset of cells expressing high levels of functional P-gp was still present in PBMC samples treated with the highest concentration of either of the PIs used in the experiments. NK cells excluding Rh123 were found even at 135 μM concentration of either indinavir or ritonavir (not shown), indicating that in certain cells, P-gp activity is present in vivo under the achievable plasma drug concentrations. The degree of P-gp inhibition in the presence of PIs is similar in both HIV-1-seronegative controls and HIV-infected subjects (not shown).

Inhibition of P-gp activity by protease inhibitors. PBMCs were stained with anti-CD3 and CD56 antibodies after 3 h of incubation in Rh123 free medium in the presence of the indicated PI concentrations. Gates were set for CD3+ (T lymphocytes) and CD3−CD56+ (NK) cells. Histograms show the number of cells with the corresponding amounts of intracellular Rh123 in the presence of increasing concentrations of either indinavir (
Discussion
PIs are an important class of drugs for the control of HIV-1 infection. PI distribution in vivo is affected by their absorption at the gastrointestinal tract, metabolism, excretion, and conjugation with plasma proteins. 51,52 Several studies have demonstrated that PIs are substrates for the P-gp efflux pump. 35,53 –55 Their absorption is directly influenced by P-glycoprotein activity in the intestinal mucosa and therefore distribution and excretion of the drugs are affected by P-gp activity. 33,56 In addition, an inverse correlation between P-gp activity and intracellular concentration of PIs has been demonstrated in vitro using human cell lines with distinct levels of P-gp expression 57 and in primary lymphocytes treated with P-gp inhibitors. 58 It has also been shown that MDR-1 genetic polymorphisms correlate with P-gp expression in PBMCs, plasma levels of PIs, and better response to HAART, as judged by immune recovery with increased peripheral CD4+ counts. 59,60 Taken together these results suggest that cells expressing high levels of P-gp could be less susceptible to the antiviral effects of PIs.
In recent years boosted PI regimens composed of low dose ritonavir with a second PI have demonstrated efficacy in clinical trials due to improved pharmacokinetics. Boosted regimens have been shown to improve the plasma levels of the second PI and to provide high-level suppression of plasma virus load. 61 Ritonavir is an inhibitor of cytochrome CYP3A4, 62 indicating that its action on improved pharmacokinetics is the result of slower drug metabolism. PIs can also inhibit P-gp-mediated efflux of other substrates such as Rh123, 63,64 thus ritonavir inhibition of P-gp may contribute to the boosting by blocking efflux of PI from the cells. In agreement with these reports, our results show that PIs are able to inhibit P-gp activity in primary PBMCs, although high drug concentrations are required to achieve full inhibition of the pump. Because these concentrations are significantly higher than the plasma drug concentration at the trough, it is concluded that the intracellular concentration of PIs at therapeutic doses are strongly influenced by P-gp in certain cellular subsets. This may result in a decrease in antiviral effects at the single cell level.
High P-gp activity is present in CD8+ T lymphocytes, NK cells, and a subset of CD4+ cells, and analysis of sequential samples from the same individual suggests that high levels of P-gp function are constitutive in these cell subsets. Our analysis of functional P-gp levels in CD4+CCR5+ lymphocytes shows a high frequency, 16–56%, of cells with high P-gp activity. Similar experiments using CD4+CXCR4+ primary lymphocytes cannot be performed reliably due to the rapid and strong upregulation of CXCR4 by most lymphocytes on culturing. When CD4+ Pgphigh cells become infected in vivo their high levels of functional P-gp might significantly reduce the intracellular concentration of PIs, resulting in suboptimal effects of the drugs and release of infectious virions. These cells could be a source of the low levels of residual HIV-1 production in patients receiving HAART, and therefore could represent a pharmacological sanctuary without a defined anatomical location. This possibility is supported by our results with HIV-1-infected sorted primary CD4+ cells treated with different concentrations of ritonavir: in these in vitro experiments, high P-gp activity decreased the cell susceptibility to the pharmacological effects of ritonavir resulting in suboptimal inhibition of HIV-1 at a drug concentration of 50 nM. Measurements of trough PI plasma levels indicate a wide range of drug concentration among different individuals. 46 –50,65 Most of the drug present in plasma are protein bound, which may result in a decrease of pharmacologically relevant drug concentration. α1-Acid glycoprotein has been identified as the main protein bound to PIs in vivo, and its presence dramatically increased the EC50 concentration of the drugs in vitro. 66 In addition, HAART-experienced patients can develop resistance to PIs, 67,68 therefore, HIV-1 strains requiring higher drug concentrations represent an additional obstacle in the context of cells with decreased susceptibility to the pharmacological effects of PIs. For these reasons, it is possible that the PI levels achieved in certain cells or tissues under present drug formulations are not sufficient to fully inhibit HIV-1.
Several reports indicate that P-gp activity is impaired in HIV-1-infected individuals 41 –43 compared to healthy controls. Our analysis of P-gp activity in PBMC samples shows no difference between HIV-1-infected individuals and healthy controls on a per-cell basis, if we measure the P-gp efflux as a percent of the initial Rh123 concentration. HIV-1-infected patients accumulate more Rh123 on loading, but display an overall similar efflux activity. Interestingly, when examining the population of CD4+CCR5+ cells ex vivo, we found that the cells with high P-gp were selectively depleted within this population in HIV-1-infected persons. This demonstrates a significant effect of HIV-1 infection on the CD4+ cells with high P-gp, which is more prominent than depletion of the total CD4+ population. Therefore, HIV-1 infection preferentially depletes CD4+CCR5+ cells with high P-gp, suggesting that either these cells are preferentially infected and/or killed by the virus, irrespective of the antiretroviral drugs used in the treatment, or that other indirect mechanisms leading to depletion are in place. The former explanation implying direct virus killing is in agreement with the phenotype of these cells as targets for HIV-1 infection. It will be important to determine whether this is a direct effect of HIV-1 on this population or whether the production/maturation/trafficking of these cells is affected.
It has been reported that high levels of P-gp expression have a protective effect against HIV-1 infection. 44,45 These data were obtained by infection of cell lines and may not reflect the situation in primary cells. In our experiments, we found no evidence of a protective role of P-gp against infection with HIV-1 in primary CD4+ lymphocytes. Our results with PBMCs infected with GFP-tagged HIV-1 molecular clones clearly demonstrate the presence of a subset of primary cells productively infected with HIV-1 having high levels of functional P-gp pump. Therefore, results suggesting a protective role of P-gp against HIV-1 infection may not apply to primary cells. In agreement with this conclusion, no correlation was found between polymorphisms in the MDR-1 gene conferring lower P-gp expression and susceptibility to HIV-1 infection. 69
Cellular factors are probably important also for antiretroviral regiments without PIs; it has been shown that MRP4 and MRP5, two members of the C family of ABC transporters, have the ability to decrease the intracellular concentration of nucleoside analog RT inhibitors. 70,71 The cellular distribution and contribution of MRP4 and MRP5 to the residual viral replication in patients receiving antiretroviral therapy remain to be determined. Our results suggest the possibility of pharmacological cellular sanctuaries resistant to the antiviral effects of PIs. The presence of these cells could be responsible for HIV-1 persistence at low or undetectable levels without the appearance of mutations conferring resistance in patients receiving HAART. New therapeutic interventions may be required to successfully eliminate those HIV-1-infected cells.
Footnotes
Acknowledgment
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Disclosure Statement
No competing financial interests exist.