
Abstract
The HIV-1 Nef protein is known to be secreted, and our group has shown that Nef is secreted from nef-transfected and HIV-1-infected cells in small exosome-like vesicles (d. 40–100 nm). The role of secreted Nef remains to be fully characterized. Thus, it is important to characterize the nature of and the mechanisms regulating Nef secretion. We hypothesized that specific structural domains on the Nef protein interact with components of the endosomal trafficking machinery, sorting Nef into multivesicular bodies (MVB) and packaging it in exosome-like vesicles. To identify those domains, a series of mutants spanning the entire nef sequence were made and cloned into the expression vector pQB1, which expresses the mutants as Nef-GFP fusion proteins. These constructs were used in transient transfection assays to identify sequences necessary for secretion of the Nef-GFP fusion protein. N-terminal domains were identified as critical for Nef-induced vesicle secretion: (1) a basic cluster of four arginine residues (aa 17, 19, 21, 22), (2) the phosphofurin acidic cluster sequence (PACS; Glu62–65), and (3) a previously uncharacterized domain spanning amino acid residues 66–70 (VGFPV), which we named the secretion modification region (SMR). Additional amino acids P25, 29GVG31, and T44 were identified in HIV-1 Nef as regulating its secretion. These residues have not been associated with other reported Nef functions. The myristoylation domain, ubiquitination lysine residues, and the C-terminal portion of Nef (aa 71–206) had no effect on secretion. A minimal HIV-1 Nef sequence, comprising the identified motifs, was sufficient for Nef-induced vesicle secretion.
Introduction
C
A large and growing body of evidence demonstrates that the Nef protein plays a key role in the pathogenesis of SIV infection. These studies show (1) primates and humans infected with nef-deleted viral strains have delayed, or no pathogenicity. 35 –39 (2) Mice and rats expressing HIV-1 nef as a sole transgene develop symptoms similar to those observed in human AIDS patients. 40 –48 (3) Nef can induce apoptosis in T cells. 49 –51 (4) A chimeric caprine arthritis-encephalitis virus (CAEV) construct expressing Nef from SIVsmm PBj14 nef was used to infect goat macrophages. 52 This led to activation and apoptosis in cocultured goat lymphocytes, even though these lymphocytes were not sensitive to infection with the virus. In a recent study, Homann et al. 53 examined HIV productive infection of human lymphoid aggregate ex vivo cultures (HLAC) from tonsil with viruses containing various Nef mutants to analyze which of Nef's activities contribute to HIV pathogenesis. 53 Their evidence suggested that Nef enhanced CD4+ T cell depletion in the absence of a significant effect on virus replication, and the Nef-dependent enhancement in depletion occurred predominantly in uninfected bystander CD4+ T cells. These reports clearly suggest that Nef markedly contributes to bystander elimination of CD4+ T cells and pathogenicity.
Most of the literature on HIV-1 Nef has focused on the endogenous functions of Nef in infected cells. However, Nef is known to be secreted 51,54,55 and has been shown to be present on the surface of infected cells. 56,57 Secreted Nef is also present in the serum of infected patients. 51 Our data, and those from a few other studies, show soluble Nef to be implicated in multiple biological activities. These include (1) stimulation of HIV transcription in promonocytic cells, 58 (2) disruption of normal hematopoiesis in bone marrow progenitor cells, 59 (3) activation of STAT1 and STAT3 in primary human monocytes/macrophages, 60,61 (4) blood–brain barrier changes, 62 and (5) induction of apoptosis in uninfected cells. 50,51,54,55 Our group has shown that secreted Nef protein can induce apoptosis in a variety of cells, including primary human T cells. This apoptotic effect of secreted Nef has been suggested to coincide with Nef's role in depletion of T cells during infection in vivo. 54,55
Our group previously reported that Nef is secreted from Nef-transfected cells in a high-molecular-weight form and vesicles are found in this high-molecular-weight differential centrifugation pellet. 63 The majority of the Nef protein was shown to be on the luminal side of these vesicles, although there was clearly Nef protein on the vesicle surface as well. Nef has been shown to be secreted from infected cells 51,64,65 and in one report Nef was found to be secreted in association with small membrane-bound vesicles. 64 It was also shown to be present in the serum of HIV-infected patients. 51 T cell lines stably expressing SIV and HIV Nef have been shown to have a dramatically altered subcellular morphology and an increase in the numbers of endosomes and lysosomes present in the cell. 66 Nef-induced intracellular vesicles were found to contain cathepsin D and LAMP2, which are characteristic features of endosomes and lysosomes. The expression of Nef also appeared to induce the intracellular accumulation of multivesicular bodies (MVBs), 67,66 which can fuse with the plasma membrane and release extracellular vesicles or exosomes. Retroviral preparations from several cell types have been shown to contain exosome-like vesicles. 68 In recent studies, it was shown that these vesicles could be separated from virion particles using an anti-acetylcholinesterase (AChE) or an anti-CD45 antibody pulldown method. 69,70 However, does Nef specifically induce vesicle secretion? Also, what are the genetic mechanisms underlying Nef-induced vesicle secretion? In this report, we find that Nef induces increases in the intra- and extracellular concentrations of vesicle-specific proteins. Furthermore, we identify key, noncontiguous and highly conserved sequences that are present in most HIV Nef proteins that promote secretion of Nef in exosome-like vesicles containing AChE and CD45. Several of the identified sequences do not overlap with residues reported previously to engage in other Nef-related interactions.
Materials and Methods
Cells and reagents
Bacterial cells
Escherichia coli STBL-2 cells (Invitrogen, Palo Alto, CA) were maintained in LB broth or LB agar (Becton, Dickinson and Company, Sparks, MD) plates at 30°C and plasmid-containing transformants were selected on LB agar plates containing ampicillin (100 μg/ml).
Mammalian cells
Jurkat CD4+ T cell lines derived from human T cell leukemia and human cutaneous T cell lymphoma cells, respectively, were obtained from the NIH AIDS Research and Reference Reagent Program (ARRRP). THP-1 and U-937 monocytic leukemia cell lines were obtained from the American Type Culture Collection (Manassas, VA). Cells were maintained in RPMI 1640 medium (Invitrogen) supplemented with streptomycin (100 U/ml), penicillin (100 U/ml),
Reagents
The following antibodies were used: (1) rabbit polyclonal anti-GFP antibody (Abcam, Inc, Cambridge, MA), (2) rabbit polyclonal anti-Nef antibody (NIH ARRRP) and murine monoclonal anti-Nef HIV-1 antibody (ImmunoDiagnostic, Inc. Woburn, MA), (3) monoclonal anti-CD45 antibody (Abcam Inc., Cambridge, MA); (4) monoclonal anti-AChE antibody (Chemicon, Temecula, CA), (5) rabbit monoclonal anti-GFP antibody (Abcam Inc., Cambridge, MA), (6) goat anti-Alix polyclonal antibody (Santa Cruz, Inc., Santa Cruz, CA), (7) monoclonal antitubulin antibody (Sigma, St. Louis, MO), (8) goat antirabbit IgG (H+L) labeled with horseradish peroxidase (HRP; Pierce, Rockford, IL), (9) camptothecin (Sigma, St. Louis, MO), and (10) donkey antigoat IgG-HRP (Santa Cruz, Inc., Santa Cruz, CA).
Construction of Nef mutants
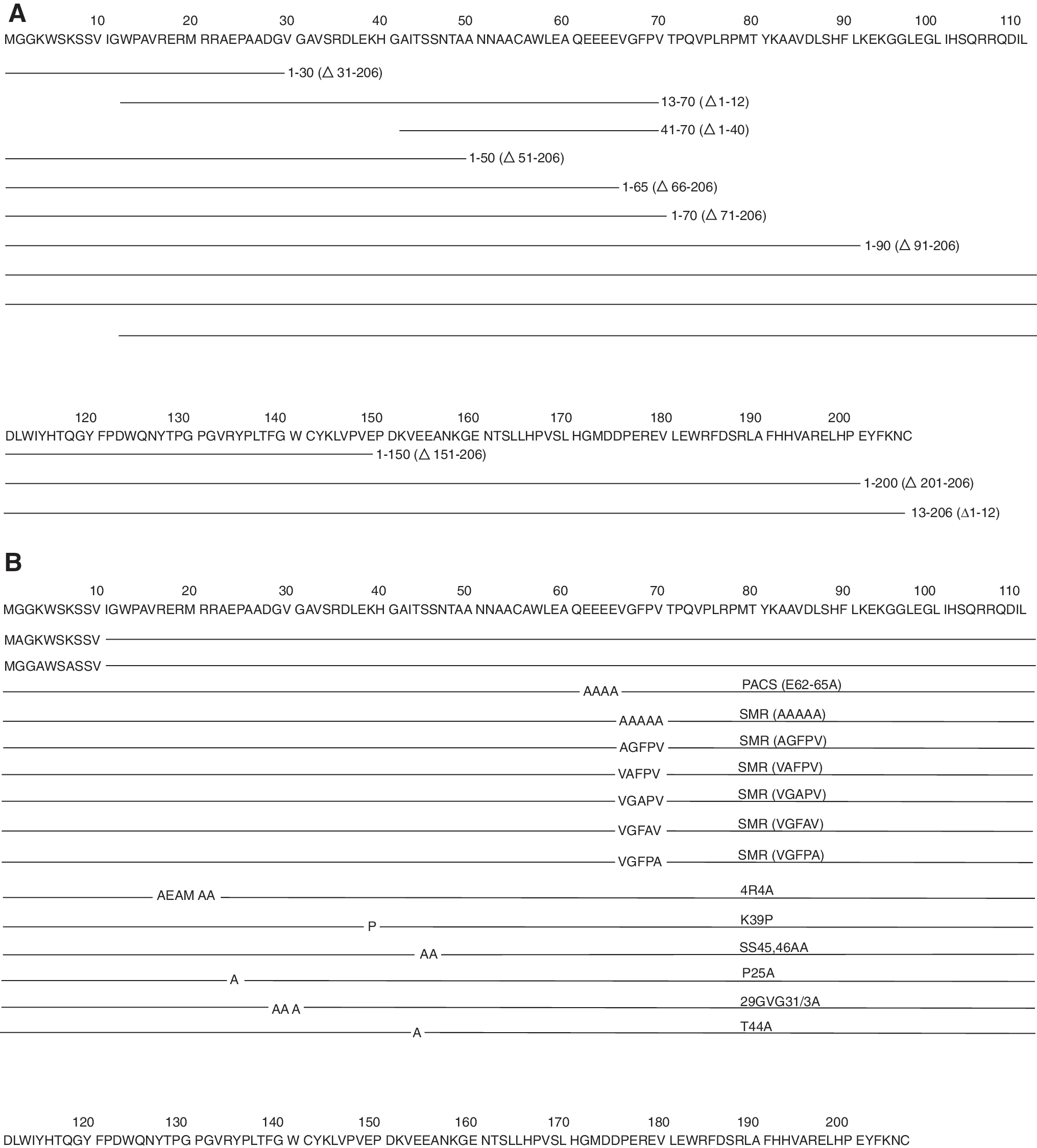
The HIV-1 NL4-3 nef construct in expression vector pQBI-Nef-GFP (Quantum Biotechnologies, Montreal, Canada) was used as a template for amplifying various Nef amplicons as well as for the subcloning of the Nef mutants to create Nef-GFP fusion constructs (Fig. 1). Nef-GFP was expressed under the control of the CMV promoter in pQBI in the various cell types tested (HEK-293, FRhK-4, Jurkat T cells and monocytes, THP-1/U937).
Schematic representation of HIV NL4-3 (
Deletion mutants of the C-terminus of HIV-1 Nef (Fig. 1A) Δ31–206, Δ51–206, Δ66–206, Δ71–206, Δ91–206, Δ151–206, and Δ201–206 were constructed by polymerase chain reaction (PCR) amplification using primers Nef-R-5798-NheI-F, Nef-R-5735-NheI-F, Nef-R-5690-NheI-F, Nef-R-5675-NheI-F, Nef-R-5615-NheI-F, Nef-R-5435-NheI-F, and Nef-R-5285-NheI-F, respectively, in combination with Nef-R-541-PvuI-R1 for PCR (see Table 1). The resulting amplicons had NheI and PvuI restriction enzyme sites on each flank. These amplicons were subsequently cloned into the NheI/PvuI sites in the pQBI vector. N-terminal deletion mutants of HIV-1 Nef Δ1–12 and Nef Δ1–40 were constructed with primers Nef13-F-SacII and Nef41-F-SacII, respectively, in combination with GFP-R-EcoRI for PCR amplification (see Table 1). The resulting amplicons had SacII and EcoRI restriction enzyme sites on each flank for subcloning into the pQBI-GFP SacII/EcoRI sites. To obtain the Δ1–12/Δ1–40 deletion mutants in the context of a full-length Nef gene, the pQBI-Nef-GFP was used as a DNA template whereas to obtain the Δ1–12/Δ1–40 in the context of the first HIV Nef 70 aa, pQBI-Nef 1–70-GFP was used as a DNA template.
All primers are from 5′ to 3′ orientation.
For the construction of HIV-1 substitution mutants (Fig. 1B) Nef 62 EEEE 65 /4AGFP, Nef 17,19,21,22 R/4AGFP, Nef 39 K/PGFP, Nef 45,46 S/AGFP, and Nef 66 VGFPV 70 GFP, primers PACS-F/PACS-R, RXRXRR-F/RXRXRR-R, XKX-F/XKX-R, XSSX-F/XSSX-R, and VGFPV-F/VGFPV-R (Table 1), respectively, were used for site-directed mutagenesis in combination with the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). A GFP expression plasmid (pQBI-GFP) was constructed by amplifying GFP using GFP-1-F-SacII/GFP-R-EcoRI primers (Table 1). The amplicon had SacII and EcoRI restriction enzyme sites on each flank for subcloning into SacII/EcoRI sites of the pQBI vector to yield pQBI-GFP.
All of the HIV Nef-GFP constructs used in this study were confirmed by sequencing of both DNA strands using CMV-846-F and GFP-1855-R primers, respectively (Table 1).
Cell transfection
HEK293 cells were grown in serum-free medium (GIBCO 293 Freestyle, Invitrogen) at 37°C to a confluence of 75–80%. Cells were trypsinized, washed, and counted before transfection with wtNef-GFP and Nef mutants using electroporation (Bio-Rad Model 1652108) as described previously. 54 Jurkat, FRhK-4, THP-1, and U937 monocytes were grown in serum-free RPMI 1640 medium and then diluted to a final concentration of 1 × 106 cells/100 μl of medium and mixed with 1 μg of plasmid DNA. The cells were transferred to electroporation cuvettes (2 mm, Bio-Rad), pulsed at 140 V (Jurkat), 130 V (FRhK-4), and 140 V (THP-1 and U937 monocytes) using a Bio-Rad Model Gene Pulser Xcell system, following the manual to select conditions. The cell/DNA solution was then centrifuged at 600 × g for 5 min, the floating dead cells were removed, and the pellet was resuspended in 1 ml of fresh media containing 5% fetal bovine serum (FBS). The cells were put in culture plates and incubated for 48 h at 37°C. Cells were collected by centrifuging at 600 × g for 5 min. The cells were mounted on a slide and the transfection efficiency was calculated by counting the green fluorescent cells using a fluorescent microscope.
Propidium iodide (PI) assay
HEK293 cells were transfected with pQBI/HIV-1 Nef mutant plasmid DNA for 48 h as described above. The cells were washed in PBS after which freshly prepared PI solution (1.25 μg/ml) was added. The cells were incubated at room temperature for 2 min and examined immediately under a microscope, with dead cells staining red.
TUNEL assay
The HEK293 cells were transfected with pQBI/HIV-1 wtNef-GFP or wtNef-RFP plasmid DNA for 48 h as described above. The cell cultures were assayed for apoptosis by TUNEL assay, by epifluorescence detection, on a computer-controlled fluorescence microscope system (Carl Zeiss, Thornwood, NY) as described previously. 71 Cells transfected with wtNef-RFP were visualized as red, whereas the TUNEL-labeled apoptotic cells were green.
Exosome isolation and purification from the transfected Jurkat cells
Cells transfected with HIV-1 wtNef-GFP (106 cells/ml, as described above) were harvested at 48 h posttransfection. The cells were removed from the culture media by centrifugation at 600 × g for 5 min. The cell-free supernatant was subjected to a second spin at 10,000 × g for 30 min to pellet the cell debris. Exosomes were collected by sequential centrifugations of this cleared supernatant at 50,000 × g for 45 min, 100,000 × g for 1 h, and 400,000 × g for 2 h at 4°C. As a negative control, culture media from a similar volume of untransfected cells were also subjected to sequential centrifugations. We also found that exosome-like vesicles could be isolated from untransfected Jurkat cells by starting with conditioned media from a larger number (2.5 × 107 cells) of cells using the same procedure.
Exosome flotation on continuous sucrose gradients
Jurkat cell cultures were transfected and distributed in 35-mm dishes (1 ml/dish) as described. For the preparation of exosomes on flotation gradients, 28 ml of untransfected Jurkat cell cultures and 14 ml of HIV-1 wtNef-GFP-transfected Jurkat cell cultures were centrifuged for 5 min at 600 × g to remove the cells. The cell pellets (see Fig. 3, lane 1) were set aside for processing (SDS–PAGE and Western blot, described below). The cell-free supernatants were then centrifuged for 10 min at 1200 × g and an aliquot (4 ml untransfected and 1 ml wtNef-GFP-transfected) of this 1200 × g clarified supernatant (see Fig. 3, lane 2) was also processed for Western blotting. The remaining clarified supernatants (24 ml untransfected and 12 ml wtNef-GFP-transfected) were subjected to sequential centrifugation for 30 min at 10,000 × g, 45 min at 50,000 × g, 60 min at 200,000 × g, and 60 min at 400,000 × g, using a Type 42.1 ultracentrifuge rotor (Beckman Instruments, Inc., Fullerton, CA). The 50,000 × g pellets were saved for Western blotting (see Fig. 3, lane 3). The 200,000 × g and 400,000 × g pellets were resuspended in 1 ml of 2.5 M sucrose, 20 mM HEPES/NaOH, pH 7.2. An aliquot (250 μl) of each sucrose suspension was centrifuged at 400,000 × g for 60 min. These samples (Fig. 3, lane 4) were set aside for Western analysis. A 10-ml linear sucrose gradient (2.0–0.25 M sucrose, 20 mM HEPES/NaOH, pH 7.2) was layered on top of the remaining 750 μl of sucrose suspension in a Beckman Ultra-Clear 14 × 95-mm tube and centrifuged at 100,000 × g for 16 h using a Type 40-Ti rotor (Beckman Instruments, Inc.). Gradient fractions (12 fractions of 750 μl) were collected, subsequently diluted 1:3 with phosphate-buffered saline (PBS), and centrifuged for 60 min at 400,000 × g using a TLA 100.4 rotor (Beckman Instruments, Inc.). The resultant pellets (gradient fractions; see Fig. 3, lanes 5–12) were set aside for Western blot analysis.
Processing of fractions for SDS–PAGE
Aliquots of the 1200 × g clarified supernatants from untransfected and wtNef-GFP-transfected cultures and the fractions from the other steps were centrifuged at 400,000 × g. The pellets were collected and lysed in 2 × SDS–PAGE sample buffer and heated at 95–100°C for 5 min. The 400,000 × g spent supernatants after differential centrifugation were processed by trichloracetic acid (TCA) and acetone precipitation. TCA was added to each supernatant to a final concentration of 15% and the precipitates were allowed to form at 4°C overnight. Precipitated proteins were collected by centrifugation at 16,000 × g for 30 min and the pellets were washed twice with ice-cold acetone and finally resuspended in 2× SDS sample buffer for analysis.
Fluorescence assays
Fluorescent plate reader assay
One hundred microliters of cell-free conditioned media was transferred to each well of a 96-well black microtiter plate (Corning Incorporated, NY). These were assayed for fluorescence on a Tecan GENEios fluorimeter (Tecan Group, Switzerland) with excitation wavelength 485 nm and emission wavelength 515 nm. Conditioned media from pQBI-GFP-transfected and untransfected cells were used as positive and negative control, respectively.
Immunoblot analysis
Cells and vesicle proteins were analyzed using Western blot analysis as described previously. 54 The cell or vesicles protein samples were separated by SDS–PAGE on a 4–20% Tris–HCl Criterion precast gel (Bio-Rad Laboratories, Hercules, CA) and electrophoretically transferred to the nitrocellulose membrane. The membrane was washed in Tris–buffered saline (TBS) for 5 min, blocked with 5% nonfat milk in TTBS (TBS with 0.1% Tween 20) for 1 h by shaking at room temperature, processed for immunoblotting using a specific first primary antibody with shaking at 4°C overnight, followed by a secondary HRP-conjugated IgG (H+L) antibody. Protein bands were detected by Western Blotting Luminol Reagent (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) followed by an exposure to photographic film (BioMax film; Fisher Scientific, Pittsburgh, PA). In some experiments, the membrane was stripped using a stripping reagent (Pierce, Rockford, IL) and used to hybridize with a different primary and secondary antibody. The X-ray films were scanned into Adobe Photoshop 5.0.2, and arranged for publication in Adobe Illustrator 10 (Adobe Systems, San Jose, CA).
Nef protein sequence alignment
The consensus Nef amino acid sequence for each HIV-1 clade (A through O) was determined by alignment of individual Nef variant sequences downloaded from the HIV Sequence Database (Los Alamos National Laboratory) using the algorithms in GENEious Pro 4.0.2 (Biomatters Ltd., Auckland, NZ). Specifically, alignments were generated using a Blosum62 Cost Matrix, with a gap opening penalty = 12 and gap extension penalty = 3. The 13 HIV-1 clade consensus sequences thus determined were then submitted for alignment in GENEious Pro, using the same parameters.
Data analysis
The numerical and graphic analyses of all data obtained were obtained through analysis using at least three repetitions of each experiment. Data were calculated and graphs were generated using SigmaPlot 10 (Systat, San Jose, CA). One-sided Student's t-test analysis was used to compare data conditions.
Results
Exosome secretion
We have previously shown that Nef is released in high-molecular-weight form from transfected cells. 63 The evidence presented in Fig. 2 shows that Nef-GFP transfected in Jurkat cells also release AChE and CD45 (exosomal marker proteins). The evidence suggests that more of these two proteins were secreted from Nef-GFP-transfected cells (Fig. 2B, lower panel set; Fig. 2C) than from untransfected cells (Fig. 2B, top panel set; Fig. 2C). Nef-GFP-transfected cells also display an increase in intracellular AChE and CD45 concomitant with AChE release (Fig. 2A, UT vs. Nef, AChE; Fig. 2C) or CD45 release (Fig. 2A UT vs. Nef, CD45; Fig. 2C) while no change in intracellular tubulin is observed (Fig. 2A, UT vs. Nef, tubulin). This clearly establishes a Nef protein-induced increase in intracellular AChE and CD45 concomitant with release of Nef, AChE, and CD45 in high-molecular-weight format. This is consistent with intracellularly expressed Nef-inducing secretion of vesicles containing Nef, AChE, and CD45.
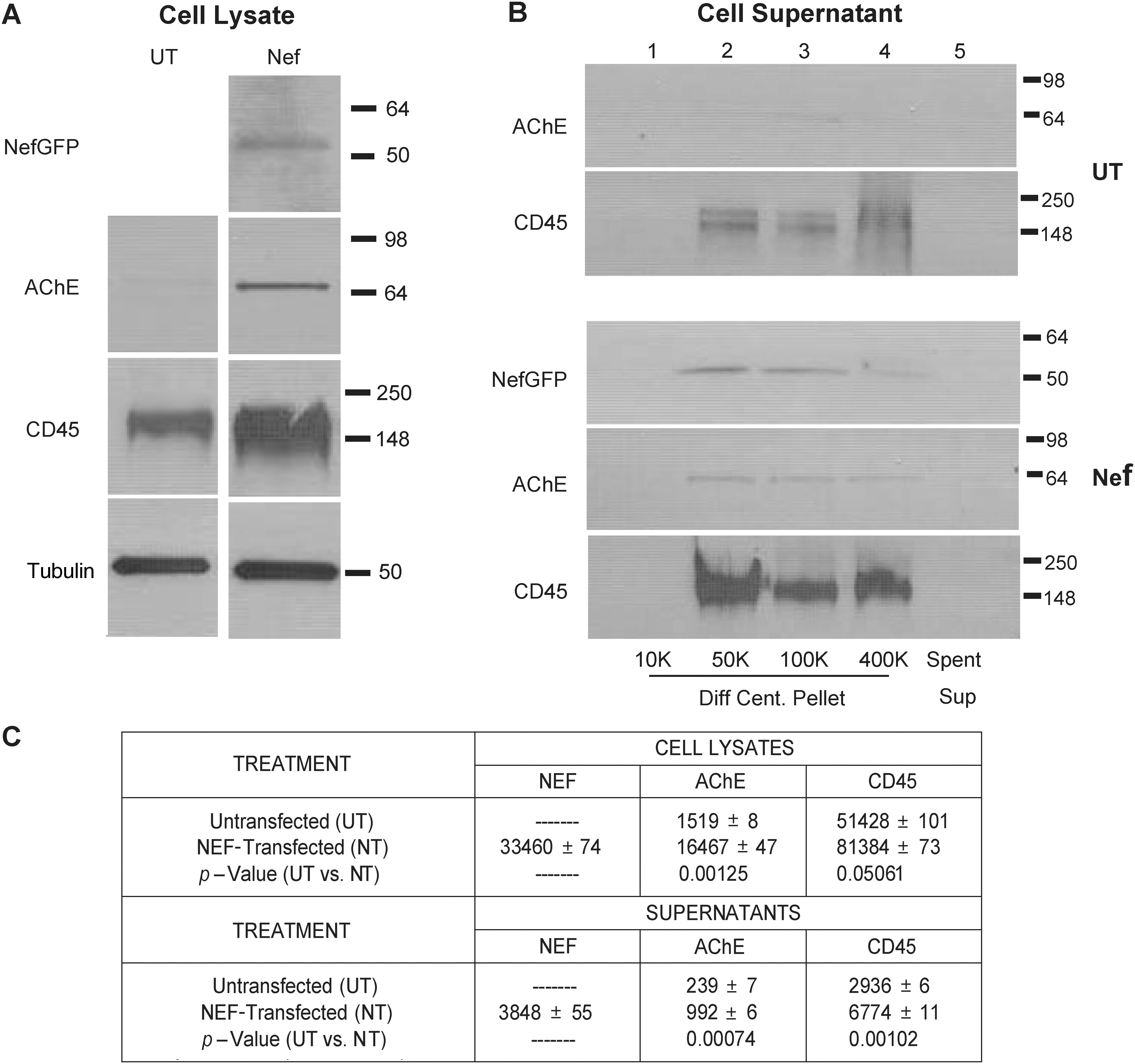
The measurement of AChE and CD45 release from untransfected and Nef-GFP-transfected Jurkat cells. Naive or HIV-1 wtNef-GFP transfection in 1 × 106 Jurkat cells for 48 h at 37°C. (
Nef protein is found in vesicular form and not in soluble form
If Nef is associated with vesicles, some fraction of the secreted material should be membrane associated. This can be demonstrated by subjecting the pelleted material from the cell supernatants to membrane flotation. Thus, Jurkat cell cultures were transfected with pQBI-Nef-GFP expressing full-length HIV-1 NL4-3 Nef, and the conditioned media from these and from untransfected cells were collected, lysed, assayed for total protein, and stored for Western analysis. Conditioned cell media were spun at 1200 × g for 10 min, the supernatant was collected, and an aliquot of this was set aside for Western analysis. The bulk of the material was subjected to differential centrifugation at 10,000 × g, 50,000 × g, 200,000 × g, and 400,000 × g and the pellets from each spin were collected. The 50,000 × g pellet was set aside to be assayed, aliquots of the 200,000 × g and 400,000 × g pellets were set aside for assay, and the bulk of these two pellets was loaded onto sucrose gradients and subjected to flotation centrifugation. Fractions from each gradient were collected and were assayed. Finally, the spent supernatant from the 400,000 × g differential centrifugation step was TCA precipitated and the pellet was resuspended in a small volume to be assayed. Each of these collected samples was assayed by SDS–PAGE and Western analysis probing for Nef, GFP, and Alix, 72 an exosomal marker. Representative Western blot images for untransfected cultures and NefGFP-expressing cultures are shown in Fig. 3A with collated densitometric measurements of multiple gradients shown in Fig. 3B.

Analysis of the vesicular nature of secreted Nef protein. Cells and conditioned media were collected from untransfected or wtNef-GFP-transfected Jurkat cultures. Culture media were processed via differential centrifugation, with spins at 1200 × g, 10,000 × g, 50,000 × g, 200,000 × g, and 400,000 × g. Both 200,000 × g and 400,000 × g pellets were subjected to sucrose gradient flotation. Cell lysates, culture medium, 50,000 × g pellets and 400,000 × g pellets and flotation gradient fractions were examined by Western blotting for Nef, GFP, and Alix. (
Several observations can be made: First, all of the Nef protein in the conditioned cell media was pelleted in the differential centrifuge steps and was found in the floated fractions of the flotation gradients (Fig. 3B). In contrast, no (soluble) Nef protein or GFP was detected in the 400,000 × g spent supernatant fraction (data not shown). Second, in the flotation gradients of pelleted vesicles, the peak band densities for Nef, GFP, and Alix were detected in gradient fractions 6–8 (Fig. 3A, lanes 7–9). Thus, our vesicle preparations floated at a sucrose density of 1.11–1.17, which is similar to flotation data reported for exosomes from B-lymphocytes. 73 Third, the amount of Alix (as measured by band densities) in all four fractions assayed was larger in the Nef-GFP-expressing cultures than in the untransfected cultures (Fig. 3B; all p values were less than 0.01). Furthermore, the difference in amount of Alix in Nef-GFP expressing vs. untransfected cell lysates and supernatants was smaller than that observed for untransfected cell lysates vs. supernatants (Fig. 3B). Finally, Nef, GFP, and Alix densitometric measurements in the differential centrifugations and the sucrose flotation gradient were found to be approximately equivalent. All this suggests that Nef increases intracellular expression of at least some specific proteins, and is released from transfected cells in vesicular form and in vesicles containing the exosomal marker Alix.
The genetics of exosome secretion
The N-terminal 70 amino acids of Nef were sufficient to induce secretion
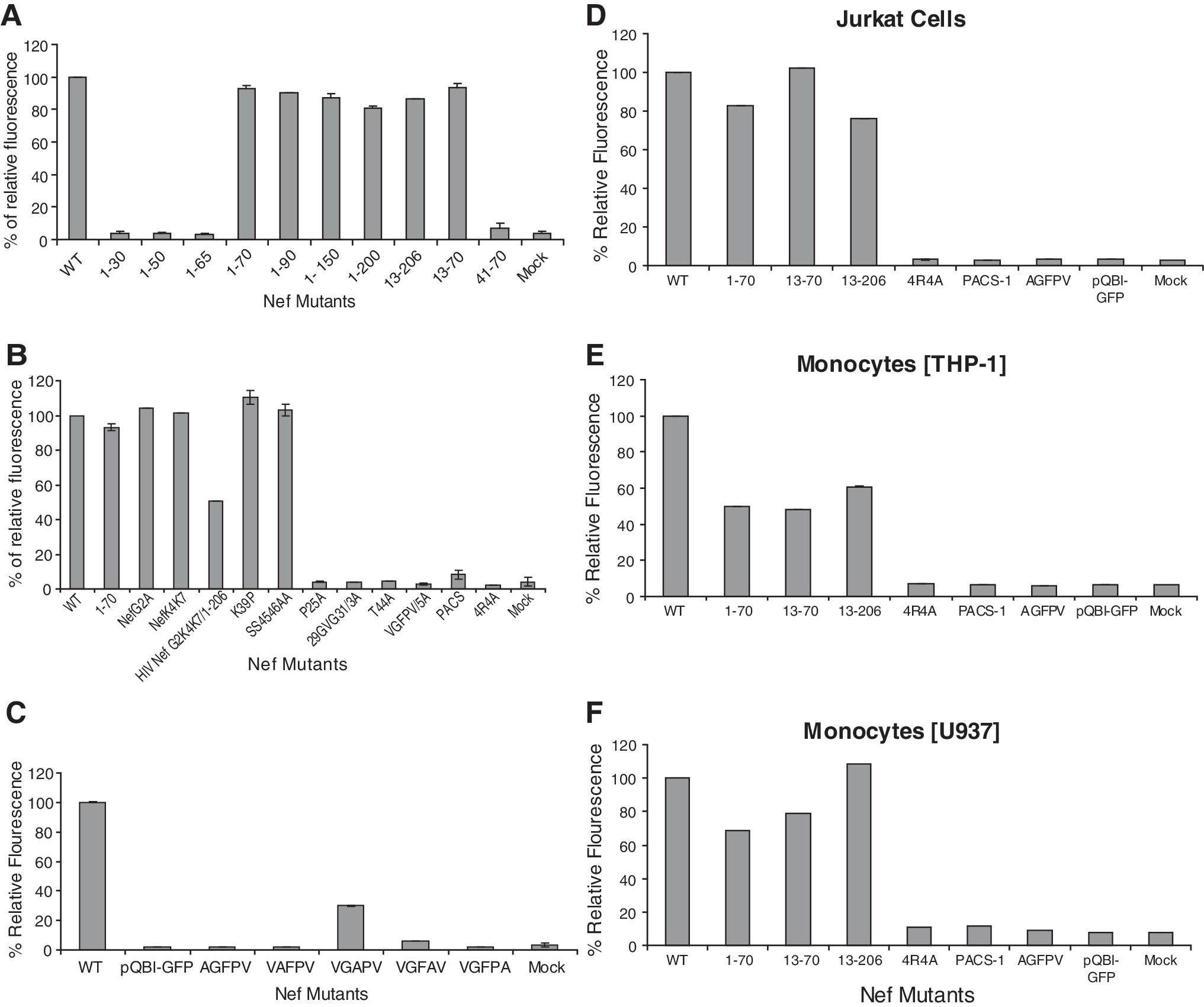
We have shown that Nef-GFP transfected into cells appears to induce release (secretion) of itself in high-molecular-weight form along with AChE and CD45. This evidence suggests that sequences or motif(s) on Nef protein actively induce and regulate this release/secretion function. The Nef motifs involved in this mechanism were initially mapped by broad truncation of the Nef gene. Most of the membrane interacting domains identified in the literature have been shown to be in the N-terminal region of the Nef protein (Fig. 4). 74 Truncation mutants deleting various lengths of the C-terminal region—NefΔ31–206GFP, NefΔ51–206GFP, NefΔ71–206GFP, NefΔ91–206GFP, NefΔ151–206GFP, and NefΔ201–206GFP (Fig. 1A)—were developed to examine their ability to induce secretion of Nef-GFP into the conditioned media using transient transfection of HEK293 cells (Fig. 5A). The clone pQBI-Nef-GFP (wt in Fig. 5A), containing the full-length HIV-I NL4-3 Nef, was used as a positive control while pQBI-GFP, containing only the GFP sequence, was used as a negative control in some experiments. Media collected from the cells transfected with pQBI-NefΔ71-206GFP (1–70 in Fig. 5A), pQBI-NefΔ91-206GFP (1–90 in Fig. 5A), pQBI-NefΔ151-206GFP (1–150 in Fig. 5A), and pQBI-NefΔ201-206GFP (1–200 in Fig. 5A) displayed fluorescence comparable to the cells transfected with full-length nef-containing plasmid. Alternatively, conditioned media from cells transfected with pQBI-NefΔ31-206GFP (1–30 in Fig. 5A) and pQBI-NefΔ51-206GFP (1–50 in Fig. 5A) displayed only background levels of fluorescence comparable to the negative control. These results showed that the N-terminal 70 aa of HIV-1 Nef were sufficient to induce secretion of the Nef-GFP protein into the conditioned media.

The structure and amino acid sequence of HIV-1 Nef. (
(
The PACS motif ( 62 –65 E) was required for Nef-induced vesicle secretion
Because the first 70 aa of Nef were sufficient for the secretion of Nef-GFP but the first 50 aa were not, it was anticipated that a secretion regulatory motif was within amino acids 50–70. There were two known motifs within this 20-amino acid region: (1) amino acids 51–61 are the apoptotic motif 55 and (2) amino acids 62–65 are the phosphofurin acidic cluster sequence (PACS) motif. 75 In previous experiments, we made deletions in the 10 amino acid apoptotic motif with no obvious effect on Nef-induced secretion. 54,55 The PACS motif has previously been shown to be required for Nef-induced downregulation of MHC-I expression and MHC-I targeting to the trans-Golgi network (TGN). 75 These effects involve binding of the Nef protein to PACS-1, a molecule that controls the TGN localization of the cellular protein furin. 76 This interaction is dependent on the cluster of four acidic amino acids, Glu62–65. 75 Furthermore, PACS-1 has been shown to be one of a family of molecules that is involved in endosomal trafficking. 76 Thus, it is conceivable that the PACS motif could be responsible for Nef-induced secretion. The PACS replacement mutant clone pQBI-Nef 62 EEEE 65 /4AGFP (PACS in Fig. 5B) was constructed by replacing the four glutamic acid residues with four alanine residues as described in Materials and Methods (Fig. 1B). As seen in Fig. 5B, conditioned media collected from cells transfected with pQBI-Nef 62 EEEE 65 /4AGFP (PACS in Fig. 5B) had only background fluorescence whereas pQBI-NefΔ71–206GFP (1–70 in Fig. 5B) had fluorescence comparable to that of pQBI-NefGFP (wt in Fig. 5B). This result suggested that the PACS region of HIV-1 Nef is a secretion regulatory motif.
The helix-1 domain but not the myristoylation domain is required for Nef secretion
Within the N-terminal 70 aa, five distinct motifs have been identified as being involved in membrane interactions (Fig. 4). These include the myristoylation region (amino acid 2), basic amino acid region 1 (BAA-1; Lys4 and Lys7), basic amino acid region 2 (BAA-2; Arg17, 19, 21, 22), which overlaps with helix-1 (Trp13–Arg21), the helix-2 (Ser34–Gly41), 77 and the plasma membrane targeting domain (PMTD, Gly41–Ala60). 74 It was possible that these or other as yet unidentified domains were also required for Nef-induced secretion. Several truncation mutants with N-terminal amino acids deleted (pQBI-Nef Δ1–12GFP and pQBI-Nef Δ1–40GFP) were constructed by deleting 1–12 aa (myristoylation region and BAA-1 were deleted) and 1–40 aa (BAA-2/helix-1 and helix-2 were deleted), respectively. No fluorescence was observed in the conditioned media collected from cultures transfected with pQBI-Nef Δ1–40GFP (41–70 in Fig. 5A), but conditioned media from pQBI-Nef Δ1–12GFP (13–206 in Fig. 5A), pQBID1–12/D71–206GFP (13–70 in Fig. 5A), and pQBI-NefΔ71–206GFP (1–70 in Fig. 5A) exhibited fluorescence intensity comparable to that of pQBI-NefGFP (wt in Fig. 5A). Cultures transfected with the mutant pQBI-NefG2A (G2A in Fig. 5B) and pQBI-NefK4K7/2A (NefK4K7 in Fig. 5B) also displayed fluorescence levels comparable to the wild-type construct, confirming the data obtained with deletion constructs. This indicated that the myristoylation domain and basic region 1 were not involved in Nef-induced secretion, whereas either the helix-1 or -2 regions, or another, as yet, unidentified domain between 13 and 41 aa was required for the secretion.
The basic amino acid motif in helix-1 is required for secretion
To determine what domain(s) between 13 and 41 aa was required for the secretion, several mutant clones were constructed (Fig. 1B). These were pQBI-Nef 17,19,21,22 R/4AGFP, in which the four basic arginines of BAA-2/helix-1 were replaced with four alanines; pQBI-Nef 39 K/PGFP, in which a proline was inserted in place of 39 K as a helix breaker in helix-2; and pQBI-Nef 45,46 S/AGFP, in which the PMTD was mutated replacing the two serines at positions 45 and 46 with two alanines. The mutations in pQBI-Nef 39 K/PGFP (Fig. 5B, K39P) and pQBI-Nef 45,46 S/AGFP (Fig. 5B, SS4546AA) had no effect on secretion of fluorescence in the conditioned media from transfected cultures comparable to that of the pQBI-NefΔ71–206GFP (Fig. 5B, 1–70) or pQBI-NefGFP (Fig. 5B, wt). Cultures transfected with pQBI-Nef 17,19,21,22 R/4AGFP (Fig. 5B, 4R4A) had significantly decreased fluorescence in the conditioned media suggesting that basic region 2 in helix-1 is important for Nef secretion.
Other previously unexplored sequences on Nef are required for secretion
To determine the minimum N-terminal sequence required for secretion we constructed a C-terminal truncation removing all amino acids after the PACS motif (pQBI-NefΔ66–206GFP; Fig. 1A). We observed a significant decrease in the fluorescence in the conditioned media from cells transfected with this construct (Fig. 5A, 1–65). This suggested that a third secretion regulatory motif lay within the amino acids 66–70 (VGFPV; see Fig. 4). Using an alanine replacement mutant clone, pQBI-Nef 66 VGFPV 70 GFP, with amino acids 66 VGFPV 70 replaced with five alanines, we observed significantly decreased fluorescence in conditioned media collected from these cultures (Fig. 5B, VGFPV/5A). Thus, this region, a domain not previously described in the literature that we named the secretion modification region (SMR), is a third region important for Nef secretion.
To characterize the SMR more fully, an individual alanine replacement analysis was performed. We developed five clones containing the full-length nef gene with nucleotides coding for one of the five amino acids of the SMR replaced with nucleotides for alanine (see Fig. 1B; 5–9). Alanine replacement mutants V66A, G67A, and V70A each displayed only background levels (Fig. 5C, AGFPV, VAFPV, VGFPA; 1.8%, 2%, 1.9%, respectively), similar to the ones measured by the pQBI-GFP-negative control (Fig. 5C, pQBI-GFP; ∼1.7%), of extracellular fluorescence in the conditioned media collected from the transfected cultures. Alanine replacement mutant P69A displayed a small but reproducible amount of extracellular fluorescence (Fig. 5C, VGFAV; ∼6%) compared to the positive control. Alanine replacement mutant F68A displayed a reduced but significant amount of extracellular fluorescence (Fig. 5C, VGAPV; ∼30%) in the conditioned media as compared to the positive control. Thus, three of the five amino acids are critical for secretion, with single mutations in any one of those three leading to complete elimination of the ability of Nef to induce secretion of itself in vesicles.
The amino acids between R22, the C-terminal amino acid in the BAA-2 motif in helix-1 and E62, the N-terminal amino acid in the PACS domain, were also screened using alanine replacement identifying several amino acids that influence secretion. These clones were developed in the full-length nef background. The pQBI-NefP25A-GFP clone (Fig. 1B, 13) displayed background amounts of extracellular fluorescence (Fig. 5B, P25A; ∼4%) in the conditioned media as compared to the positive control. pQBI-Nef 29 GVG 31 3A (Fig. 1B, 14) and pQBI-NefT44A-GFP clone (Fig. 1B, 15) also displayed background amounts of extracellular fluorescence (Fig. 5B, 29GVG31/3A, 4%; T44A, 4% respectively).
These domains are relevant in other cell lineages
The initial secretion analysis described above was performed in HEK293 cells. These cells are easily transfectable and do not normally secret vesicles. Thus, they are optimal for viewing secretion and identifying changes in the secretion ability although not a normal target for viral infection. More appropriate would be Nef secretion analysis of these constructs in either lymphocytic or monocytic cell lines as these lineages are targets of HIV infection. Specific Nef mutants described above were analyzed in a lymphocytic cell line (Jurkat cells) and in two monocytic lines (THP-1 and U937 cells; Fig. 5). The pQBI-Nef 17,19,21,22 R/4AGFP mutant clone (BAA-2 region knockdown), the pQBI-Nef 62 EEEE 65 /4AGFP mutant clone (PACS region knockdown), and the pQBI-NefV65AGFP mutant clone (SMR region substitution mutation knockdown) all displayed extracellular fluorescence levels in lymphocytic and monocytic cells similar to those observed in HEK293 cells. There was some variation in the extracellular fluorescence levels of the truncation mutant's transfected in lymphocytic (Fig. 5D, 1–70, 13–70, 13–206) and monocytic cell lines (Fig. 5E or F, 1–70, 13–70, 13–206) relative to each other or to HEK293 cells (Fig. 5A, 1–70, 13–70, 13–206). However, the variations observed were not significant and the trend for each of these truncation mutants was for them to display wild-type or close to wild-type levels of fluorescence.
Phylogenetic analysis across HIV clades
The genetic analysis of Nef secretion was performed using HIV-1 NL4-3 Nef. A logical next step was to determine the conservation of the identified secretion domains across HIV B clade viruses and across the other HIV-1 clades uncovering the relative importance of these domains. An analysis of that region of Nef involved in secretion (amino acids 1–70) demonstrates significant sequence conservation within the secretion domains across all HIV-1 clades (Fig. 6). Interestingly, the SMR domain, which was always found contiguous to and C-terminal of the PACS domain, displayed 100% sequence conservation across all the HIV clades suggesting the importance of these sequences.

The alignment of the PACS/SMR region of HIV-1 Nef. Amino acid consensus sequences for 13 HIV-1 subtypes were determined as described in Materials and Methods. The PACS-SMR consensus sequences were then aligned to illustrate the degree of homology in these required secretion domains of Nef. Dashes (-) indicate gaps inserted to facilitate the alignment.
HIV Nef expressed in cells is not toxic/apoptotic to transfected cells
One alternative explanation of the effects being observed is that endogenous Nef protein causes toxicity to the cells in which it is expressed, leading to those cells releasing Nef protein in apoptotic microvesicles or microparticles. Prior studies of cells releasing putative exosomes have shown that cells in the early stages of apoptosis release membrane vesicles that are very similar to vesicles released by healthy cells (e.g., exosomes). 72,78 However, the protein composition of the apoptotic vesicles was different from that of the exosomal vesicles. For example, the apoptotic vesicles contained large amounts of histones as opposed to little or no histone protein found in the exosomal vesicles.
We have already shown that soluble recombinant Nef (rNef ) protein and the conditioned supernatant from Nef-transfected cells are apoptotic to naive cells expressing CXCR4. 54 Thus, it is possible that these Nef-containing vesicles represent apoptotic vesicles. We examined the cells transfected with the various Nef-GFP constructs for cell death and apoptosis (Fig. 7) and the supernatant/vesicles released from the Nef-transfected cells for histone content in the vesicles, a marker of apoptotic vesicles (see Fig 8).

HIV Nef expressed in cells is not toxic/apoptotic to transfected cells. HIV-1 Nef–GFP mutants were transfected into HEK293 cells at 37°C for 48 h. Subsequently, the cultures were stained with propidium iodide (PI) to visualize the nucleus. Finally, a comparative morphological examination of the individual cells in these cultures was performed to determine whether and how much cytotoxicity or apoptosis was observed in the transfected cells. (

Nef-induced vesicles do not display attributes of apoptotic vesicles. HIV-1 wtNef-GFP and Nef-GFP mutants were transfected into HEK293 cells. (
HEK293 cells were transfected with specific Nef constructs described above, and the cell populations were stained with PI. These cells were analyzed for GFP fluorescence (NefGFP expression), PI fluorescence (necrotic cells hallmark of cell death), and coincidence of PI and GFP (dying cells expressing Nef ) in the cells (Fig. 7A). Endogenously expressed GFP fluorescence, a measure of Nef expression, for all treatments ranged between 70% and 80% and did not vary significantly. PI fluorescence, a measure of cell death, varied from 3% (pQBI-GFP; Fig. 7A, PI measure) in the negative control and the Nef mutants to ∼12% (pQBI-NefGFP; Fig. 7A, PI measure) in the transfections with wtNef-GFP. Thus, wtNef-GFP protein expressed within the cells does increase the amount of cell death by about 4-fold with about half of that cell death occurring in the transfected cells (see Fig. 7A, wtNef-GFP, GFP/PI overlay measure). However, the total amount of cell death remained modest.
HEK293 cells were transfected with wild-type pQBI-Nef-RFP and then TUNEL labeled for detection or earlier signs of apoptosis in the form of DNA fragmentation. These cells were analyzed for RFP fluorescence (Nef-RFP expression), TUNEL (apoptosis), and the coincidence of RFP and TUNEL (apoptotic cells expressing Nef ) in the cells (Fig. 7B). Endogenously expressed RFP fluorescence, a measure of Nef expression, for all treatments ranged between 75% and 80% and did not vary significantly. FITC fluorescence, in TUNEL-labeled apoptotic cells, ranged from 2% (pQBI-RFP, Fig. 7B, RFP measure) in the negative control to ∼12% (pQBI-Nef-RFP, TUNEL measure) in the transfections with wtNef. Again, wtNef protein expressed within cells increased the amount of apoptosis by about 6-fold with half of that apoptosis occurring in the transfected cells (see Fig. 7B, wtNef-RFP, RFP/TUNEL overlay measure). Again, the total amount of cell death in the population was very modest.
Thus, evidence for direct and indirect induction of apoptosis was present but minimal. The next question we needed to address was whether transfected cells released histone-containing apoptotic vesicles into the conditioned supernatant. To examine this, HEK293 cells were either treated with camptothecin, an apoptosis-inducing factor (Fig. 8A, lanes 1 and 2), or transfected with pQBI-NefGFP (Fig. 8A, lanes 3 and 4), pQBI-Nef 66 AGFPV 70 GFP (Fig. 8A, lanes 5 and 6), or pQBI-GFP (Fig. 8A, lanes 7 and 8). The 48-h cultures were harvested for the conditioned media and the cell lysates. The conditioned media from each treatment were subjected to differential centrifugation with four sequential centrifugation steps of 300 × g, 1200 × g, 10,000 × g, and finally 130,000 × g. A silver-stained SDS–PAGE analysis of the cell lysates (Fig. 8A, lanes 1, 3, 5, and 7) and 130,000 × g pellets (Fig. 8A, lanes 2, 4, 6, and 8) was examined for the protein composition of those two fractions. The banding pattern in the camptothecin-treated cells (Fig. 8A, lanes 1 and 2) was distinct from the other three transfection treatments (Fig. 8A, lanes 3–8).
To specifically look at the histones in these treatment conditions, SDS–PAGE analyses of the cell lysate of each treatment and the pellets from each centrifugation step were screened by Western analysis (Fig. 8B). This was done with (1) a histone polyclonal antibody (Fig. 8B, first panel set) to screen and quantify histones, (2) a GFP antibody (Fig. 8B, second panel set), and (3) an HIV-1 Nef antibody (Fig. 8B, third panel set). The camptothecin-treated cells (Fig. 8B, histone set, panel one) displayed a histone band in both the cell lysate as well as in all four differential centrifugation-generated pellets as expected following camptothecin-induced apoptosis: histones were detected in both the cell lysates and vesicles released in the supernatants. In comparison, HEK293 transfected with pQBI expressing wtNef, SMR mutated Nef, or untransfected control, histone bands are detected only in the cell lysates and in the low-speed centrifugations (300 × g and 1200 × g) in which cellular debris is normally pelleted. This suggests that Nef transfection does not result in significant release of apoptotic histone-containing vesicles. The transfected and wtNef-GFP-expressing cultures analyzed by GFP antibodies (Fig. 8B, GFP set, panel two) or by Nef antibodies (Fig. 8B, Nef set, panel two) display Nef-GFP protein in both the cell lysate as well as in all four differential centrifugation conditions. This indicates that Nef is there in a high-molecular-weight format indicative of Nef-containing vesicles.
The evidence suggests that despite finding an increased (but small total) amount of cell death/apoptosis in the Nef-transfected cells, the vesicles released from these cultures have very little if any histones in them, suggesting a morphology distinct from apoptotic vesicles. Alternatively, they do have Nef-GFP in them, suggesting that the Nef-containing vesicles may be exosomes.
The effect of Nef mutants was not due to variable expression
The effects observed in the various mutants could be due to variation in the ability of each clone to express the resultant fusion protein and not due to differences in their ability to secrete the fusion protein. We addressed this issue by examining the expression pattern of untransfected and transfected HEK293 cells by Western analysis of whole cell extracts probed with anti-Nef antibody (Fig. 9A) or anti-GFP antibody (Fig. 9B). Cultures were transfected with pQBI-Nef-GFP (Nef-GFP; Fig. 9, lane 1), pQBI-GFP (GFP; Fig. 9, lane 2), pQBI-Nef 62 EEEE 65 /4AGFP (PACS; Fig. 9, lane 3), pQBI-NefV66/A (SMR AGFPV, Fig. 9, lane 4), pQBI-Nef 17,19,21,22 R/4AGFP (Basic Region 2, Fig. 9, lane 5), or untransfected (Fig. 9, lane 6). Figure 9D is the densitometric analysis of Fig. 9A–C. The Nef and GFP band densities in the mutant expressing cultures are similar (Fig. 9D). Alternatively, a significant difference was observed in the band densities of wtNef-GFP-expressing cells vs. the Nef mutant-expressing cells (Fig. 9D, wtNef-GFP vs. all others). This suggests that NefGFP protein made and released in the wtNef-GFP-expressing cells accumulates within the mutant Nef-GFP-expressing cells.
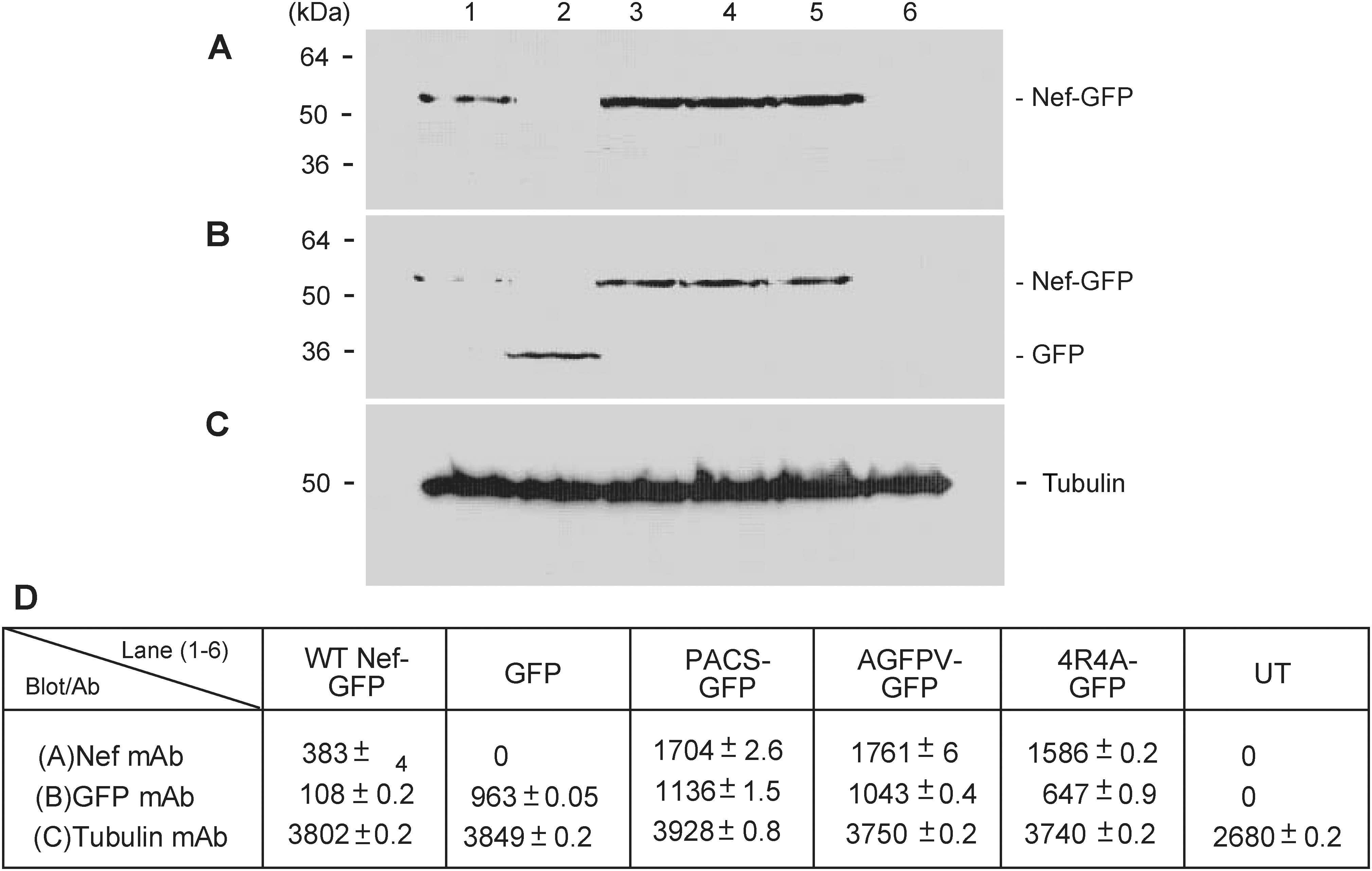
The effect of Nef mutants is not due to variable transfection/expression efficiencies. (
Discussion
In earlier studies it was shown that T cell lines stably expressing SIV or HIV Nef had dramatically altered subcellular morphology. 66 These cells displayed increased numbers of endosomes and lysosomes as well as increased intracellular accumulation of MVBs, 66 which are the precursors of exosomal vesicles. 79 Exosomes are derived from the endosomal compartment and are characterized by specific endosomal and plasma membrane proteins (e.g., AChE, 80 CD45, 81 Alix 72 ). In a previous study from our group, Campbell et al. 63 found that intracellular Nef expression causes an increase in intracellular vesicular bodies that morphologically resemble MVBs.
Our current studies comparing untransfected and Nef-transfected cells have uncovered an increase in intracellular expression of at least a subset of cellular proteins such as Alix, AChE, and CD45 with no change in other proteins (e.g., tubulin). Expression of wtNef also caused an increase in the amount of extracellular release of Alix, AChE, and CD45 into the culture medium concomitant with secretion of Nef. This increased amount of wtNef-induced extracellular protein was found exclusively contained within a vesicular format with no detectable soluble protein. On examination using sucrose flotation gradients, the data on the Nef-containing vesicles from Nef-GFP expressing cells, as well as that for vesicles from untransfected Jurkat cells, were similar to flotation data reported for exosomes from B-lymphocytes by Raposo et al. 73 Finally, we found that within this extracellular vesicular format, Nef protein was associated with proteins normally found in exosomes (i.e., Alix, CD45, and AChE). Thus, intracellular expression of Nef correlates with an increase in the numbers and concentration of intracellular exosomal markers: endosomes, lysosomes, and MVBs. Furthermore, this was concomitant with increases in extracellular Nef, Alix, AChE, and CD45, within a vesicular format that closely resembles exosomes.
Alternatively, the vesicles observed in this study could be microparticles or microvesicles. These are vesicles that are somewhat larger than but similar to exosomes. They are released from activated or apoptotic cells following the disturbance of membrane phospholipid asymmetry. Changes in phosphatidylserine (PS) and cytoskeleton degradation are followed by MP release through shedding or blebbing of the plasma membrane. 82,83 Our studies have shown the vesicles released by Nef-transfected cells display endosomal/plasma membrane proteins, which are a hallmark of exosomes. Moreover, these vesicles lack large concentrations of histones, which distinguishes them from apoptotic microparticles. 72,78 Thus, one likely interpretation of the combined evidence is that Nef protein upregulates the exosomal machinery through increased intracellular expression of exosomal proteins leading to increases in MVBs with concomitant insertion of itself (Nef ) into the MVBs. Ultimately, this leads to increased vesicle secretion into the extracellular milieu as exosomes. However, it is unclear how Nef causes (1) increased cellular expression, (2) increased secretion of vesicles, and (3) insertion of itself into the secreted vesicles.
We have developed clear evidence that specific sequences or motif(s) on the Nef protein actively regulate this vesicle release/secretion process. The N-terminal 70 amino acids of HIV-1 Nef were found to be necessary and sufficient to induce vesicle secretion. Within that region, we identified three domains as important for Nef-induced vesicle secretion: (1) an arginine-rich basic domain (aa 17–22), (2) the PACS motif, and (3) an SMR domain (65VGFPV70), which has not previously been identified with any function. These three domains are each required for Nef-induced vesicle secretion. Additionally, we identified small amino acid sequences in HIV-1 Nef that are required for Nef secretion and have never been associated with other Nef functions. These include P25, 29GVG31, and T44 (Fig. 5). The evidence suggests that changes in at least some of these domains do not simply cause major structural rearrangements in the Nef protein but identify and modify a functional domain (i.e., a domain allowing interaction with cellular factors). For example, the amino acid substitutions made in the SMR domain were relatively conservative and should not have led to loss of function through protein misfolding. Finally, evidence presented show that Nef mutations leading to elimination of vesicular secretion do not change (reduce) the Nef-GFP expression patterns but in fact lead to concomitant intracellular accumulation of Nef and GFP. Thus, measured variations in GFP levels in the conditioned culture supernatants were not due to Nef-GFP expression variations in the different mutant clones but to loss of the Nef-induced secretion ability.
Myristoylation of Nef has been thought to be required for all of its known activities 84 including the pathogenic phenotype of HIV as mutation of the glycine residue at the N-terminus abolishes Nef's ability to downregulate CD4, MHC, activate Pak kinase, and enhance infectivity. 85,86 Several studies have shown that the G2 myristoylation site is important but not the sole requirement for membrane association. Giese et al. 74 found that mutation in the G2 myristoylation site, the lysines at K4 and K7, the tryptophans at W5 and W13, and the arginine (R4)-rich region from amino acids 17–21, all had some effect on membrane association. The maximal effect was observed when the lysine and arginine mutations were combined. 74 They further suggested another site between aa 40 and 61 that also affects membrane association. Interestingly they found that the lysines were important for incorporation of Nef into lipid rafts. Bentham et al., 87 using a biochemical assay for membrane association, showed that the myristoylation was not required for membrane association. Fackler et al. 88 found that the G2 myristoylation site was not always required for Nef's presence in the viral particle but varied by virus variant. Thus, evidence from previous studies supports G2 myristoylation as important along with other domains in membrane association. This is in agreement with evidence from studies of other myristoylated proteins showing that the myristoylation site is not enough to provide a stable membrane association. 89,90 A set of basic amino acids downstream of the myristoylation site has been postulated to increase membrane stability and possibly to provide membrane specificity. 87,88,91 Fackler et al. 88 and Giese et al. 74 found that the R4 and K2 basic amino acids downstream of the G2 myristoylation site were dispensable for biological activity in HIV-1-infected lymphocytes suggesting further membrane-binding sites.
In this study we found that the N-terminal 70 amino acids were sufficient for induction of secretion in several cell lines including HEK293, Jurkat, and THP1 cells. Clearly, induction of the endosomal trafficking pathway and incorporation of Nef into exosomal vesicles require membrane association indicating all the membrane association signals should be in that first 70 amino acids. Interestingly, we could delete the first 13 amino acids, which includes the myristoylation site (G2), and basic region 1 (K4W5K7W13), and still get the maximal secretion function. Alternatively, mutation of the arginines in the basic region 2 (R4) eliminated Nef-induced secretion even in the presence of a conserved myristoylation site. Thus, whereas the N- terminal 13 myristoylation and basic amino acids are dispensable for secretion, the basic region 2 (R4) is required for secretion. Furthermore, this suggests that the membrane diffusion phenotype observed by others 74 appears to be separable from the secretion process. As it is clear that vesicle secretion requires membrane association, this evidence hints at targeting of specific membranes.
As discussed above, Giese et al. 74 postulated another membrane-targeting domain(s) downsteam of the arginine-rich domain (basic region 2/R4). In fact, we did identify sequences directly downstream of the arginine-rich domain of HIV-1 Nef that were required for secretion. P25, 29GVG31, and T43 were also found to be required for Nef-induced secretion. Interestingly, T43 falls within the HIV-1 Nef aa 40–61 region, which Giese et al. 74 suggested contained another membrane-targeting domain.
We also identified sequences further downstream of the arginine-rich domain that were required for secretion. The PACS domain, aa 61–64, was found to be critical for Nef-induced secretion. The PACS domain has been found to be involved in interactions with adapter proteins involved in endosomal trafficking in other cellular proteins. 92 This specific domain in Nef has been postulated to be involved in linking Nef with these adapter proteins involved in MHC downregulation and thus the endosomal trafficking pathway through the PACS protein. PACS domain involvement appears to tie Nef-induced vesicle secretion to the endosomal trafficking pathway. This evidence fits our postulate that these vesicles are exosomes. Finally, we identified another domain critical for Nef-induced secretion immediately downsteam of the PACS domain but upstream of the SH3 domain. Because this domain had not previously been associated with any known Nef biological function, we named it the secretion modification region (SMR).
Our initial genetic analysis was done in a cell type (HEK293) that is easily transfectable, and in which we could observe the examined phenotype (secretion) easily. Furthermore, the nef gene used to generate our constructs originated from a standard laboratory strain. For secretion to be important in the virus life cycle or for virus-driven pathogenesis, these domains should be equally relevant in cells the virus infects and/or depletes. Furthermore, HIV has many different variants and a number of clades that all cause the same pathogenic effect in humans. Thus, obvious questions arose as to (1) whether these Nef domains were equally relevant for secretion in other cell types, specifically those of hematopoietic origin, and (2) whether these domains were observed in other HIV variants and clades. Those domains found to be important for HIV-1 Nef-induced secretion in HEK293 cells were similarly important for HIV-1 Nef secretion in a lymphocytic cell line (Jurkat) and in a monocytic/macrophage cell line (THP-1). There was some variation in the relevance of the N-terminal G2K4K7 domain in that substitution mutations in that domain had only a small effect in HEK293 cells while causing loss of secretion function in the more relevant cell types. However, on removal of those sequences (aa 1–13), that truncation mutant was equally functional for secretion in both cell types. We postulate that the substitution mutations cause structural change(s) in the N-terminal domain, not allowing it to interact with an unknown factor, resulting in no new Nef vesicle formation. Alternatively, removing those amino acid sequences eliminates that constraint on the N-terminal domain allowing other domain(s) (e.g., BAA-2) to interact with the unknown factor and drive Nef vesicle formation and secretion. In support of this, we have found that introduction of new amino acid sequences onto the N-terminus of Nef also causes elimination of Nef's ability to induce Nef vesicle secretion (unpublished evidence).
A phylogenetic analysis of HIV-1 Nef amino acids 1–70 intra-B-clade and across all HIV-1 clades found that the secretion domains are highly conserved with the newly identified regions (P25; 29GVG31; T44; SMR) being very highly conserved. The SMR was 100% conserved across all HIV-1 clades (Fig. 6). This evidence indicates the relevance of these domains, particularly in a virus that displays high sequence variability. Interestingly, domain conservation was also found to apply when the N-terminal sequences of HIV-1 and SIV were compared (data not shown). Although most of the Nef secretion regulatory sequences were found in the N-terminal 102 amino acids of SIV Nef, the three functional motifs in association with Nef secretion in high-molecular-weight form are very similar to HIV and comprise two BAA regions, a PACS domain and an SMR-like region located immediately downstream of the PACS. We have created a similar set of mutants in SIV Nef to determine the functionality of those regions and found that they regulate SIV Nef secretion in a way similar to that of HIV Nef (unpublished observations).
The benefit of vesicular secretion of Nef to HIV or SIV remains to be elucidated. However, the high mutation rate of lentiviruses in vivo contrasted with the high level of conservation of the Nef motifs associated with the secretion mechanism reported herein clearly points to a key mechanism that has withstood millions of in vivo passages (Fig. 6). We suggest that this secretion function is critical for the virus to replicate and to be successfully transmitted either via some undiscovered viral property or as a mechanism evolved by the virus to counter effective antiviral immune defenses. Such a mechanism has been proposed and has been supported by a body of evidence describing the physiologic processes that lead to immune privilege during pregnancy 93 and that are hijacked, leading to immune suppression allowing tumor growth. 94 In both cases, release of FasL-containing exosomes has been shown to be one mechanism by which the placenta and tumors promote a state of immune privilege/immune suppression. 93 The proapoptotic effect observed in bystander, CXCR4-expressing, naive T cells is readily understood. However, a direct link between the secretion mechanism and the HIV replication advantage in vivo is still to be deciphered. Our study represents an initial step in the elucidation of this potentially key viral function, and paves the way for future investigations into the pathogenic potential of this mechanism.
Note Added in Proof
Since this article was accepted, Muratori et al. (Cell Host Microbe 2009;6(3):218--230) have showed massive secretion of Nef-containing vesicles from the cells infected with nef+ NL43 HIV strain. This vesicle secretion was strictly Nef dependent as it was not seen in the cells infected with Δnef-NL43 strain. The authors also showed that Nef could effectively be transferred to the bystander cells through the Nef-containing vesicles and resulted in Nef-associated signaling activities to bystander cells. Based on their findings, the authors suggest that microvesicle release could potentially explain the effects observed in bystander cells. This observation was further supported by the recent publication of Lenassi et al. (Traffic 2010;11(1):110--122). The authors demonstrated that the expression of Nef not only augmented the production of the exosomes from the cells but also resulted in the packaging of Nef into these vesicles which, upon contact, lead to an activation-induced cell death in the resting CD4+ T lymphocytes (bystander cells). These studies also confirmed the earlier findings of Bond's lab on Nef-induced apoptosis in CXCR4 positive cells. Our present work supports the concept of Nef-containing vesicle release and provides Nef structural domains required for generation/release of those Nef-containing vesicles.
Footnotes
Acknowledgments
This work and the researchers were supported by NIH/NIGMS/MBRS (Grant 58268), NIH/NCRR/RCMI (Grant G12-RR03034), and Georgia Research Alliance funding Grant GRA.VAC08.W. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Grant C06 RR18386 from the NIH/NCRR. The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: HEK 293 cells and anti-Nef polyclonal antibody. We thank the MSM RCMI Gene Profiling Core/Molecular Genetics laboratory and Mrs. Qi Yang for sequencing the many clones that were developed as part of this study. S.A. Ali and M-B. Huang contributed equally to this work.
Author Disclosure Statement
No competing financial interests exist.