
Abstract
We cloned and sequenced gp41 HIV-1 from plasma of AIDS patients under HAART and T-20 (enfuvirtide, Fuzeon) therapy and revealed several T-20 resistance-associated mutations. Two mutations, a single V38A and a double N43T-N44K were the most frequent; however, they were not found together in one clone. We anticipated that simultaneous mutations of these three residues might play a vital role in the viral life cycle. To address this problem, we introduced N43T-N44K and V38M + N43T–N44K substitutions to a cloned gp41 and introduced modified gp41 into the pNL4-3 molecular clone. HEK293T cells were transfected with the obtained vectors and released viruses were examined for reverse transcriptase (RT) activity, infectivity on reporter TZM-bl cells, and in Western blotting. Nearly equal RT activity was demonstrated in viruses with and without mutations. However, viruses with the V38M + N43T–N44K mutations were not infectious and, as shown by Western blotting, gPr160 cleavage was impaired. These data suggest that V38M + N43T–N44K mutations perturbed the natural conformation of gPr160 in a way that access of furin to the cleavage site (REKR) was blocked. Therefore, the residues V38 + N43–N44 retain the gPr160 conformation in proximity to the furin cleavage site and, as a consequence, are critical for virus infectivity. These data may explain why viruses with V38M + N43T–N44K mutations were not previously detected in the plasma of T-20-experienced patients.
A
The HIV-1 genome is variable and the env gene is the most inconsistent element. The HIV-1 envelope complex is synthesized as a precursor, gPr160, 1,2 that is cleaved in the rough endoplasmic reticulum-Golgi complex 3,4 by a furin-like protease at the consensus site (REKR decreased) behind the carboxy-terminal arginine. 5 –7 Cleavage of gPr160 creates the surface–gp120 (SU) and the transmembrane gp41 (TM) glycoproteins. 8 gp120 and gp41 interact in noncovalent way and exist as trimeric complexes on the plasma membrane. 9,10 gp120 determines virus tropism and gp41 is responsible for virus-to-cell fusion. Several domains of the gp41, including the hydrophobic fusion domain (FD), and helical heptad repeats 1 and 2 (HR1, HR2) are directly engaged in this process. 11 Disruption of the gPr160 cleavage by site-directed mutagenesis 12 or inhibition of gPr160 processing with polyarginine 13 resulted in noninfectious virus particles.
Sequence variability and dense glycosylation of ENV allow the virus to escape out of immune pressure. 14,15 These properties of the virus significantly complicated antiviral therapy. However, despite prominent polymorphism, several fragments including parts of the gp41 HR1 and HR2 are rather conservative. These regions in gp41 are targeted by a family of antiviral drugs, the fusion inhibitors.
T-20 (enfuvirtide, Fuzeon) is a synthetic 36-residue peptide corresponding to residues 127–162 of the HR2; it represents the first generation of fusion inhibitors. 16 –18 T-20 is a class 1 fusion inhibitor 19 that interfers with the HR1 region of the gp41 and prevents HR1 interaction with HR2. As a consequence of that, the six-helix bundle configuration and lipid mixing do not occur. However, in vitro assays, 20 as well as numerous in vivo data, demonstrated that T-20 therapy resulted in the rapid appearance of antidrug resistance and most of the resistance-associated mutations are localized in HR1 of gp41 between amino acids 36 and 45. 21 –25 It is noteworthy that viruses with resistance to T-20 were also found in therapy-naive patients. 26 –28
Earlier, by clonal analysis, we revealed several T-20 resistance-associated mutations in HIV-1 (clade B)-infected AIDS patients under combined HAART–T-20 therapy. 28 As expected, all mutations were mapped to the HR1 36–45 amino acids region and most of them were either single V38M or double N43T–N44K. It has been shown that both mutations alone are rather frequent in T-20-experienced patients. 21,29,30 In the absence of the drug the viruses with these mutations demonstrated reduced replication rates (less fit) compared to the wild type 29 and a pause in T-20 therapy resulted in a recovery of the T-20-sensitive genotype. 25,28,31
Since both V38M + N43T–N44K mutations were not revealed among cloned env sequences and were also not reported by other groups, we suggested that simultaneous mutations might be vital for the virus. To confirm this suggestion, we therefore introduced two N43T–N44K and three V38M + N43T–N44K substitutions into the reverse transcriptase polymerase chain reaction (RT-PCR) env amplicon (primers 7487F–5′ aacatgtggcaggaagtaggaaaag; 8490R–5′ tcgtcccagataagtgctaa; GenBank accession number AF324493). The env amplicon was obtained from a plasma sample of the therapy-naive HIV-1 clade B-infected seroconverter 01-0209 (GenBank accession number EF192132) and it was cloned into a pCR2.1-TOPO vector (Invitrogene). The substitutions were made using the QuickChange multi-Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). Clones with and without substitutions (control) were further cleaved by BamH1–BsaB1 restriction enzymes and the inserts were phenotyped with a pNL4-3 infectious molecular clone. Plasmids were isolated using the GeneJet Plasmid Miniprep kit (Fermentas Life Sciences, Hanover, MD) and substitutions were verified by sequencing. The plasmid without substitution in the insert is further referred to as “p209” and plasmids with substitutions as “p209
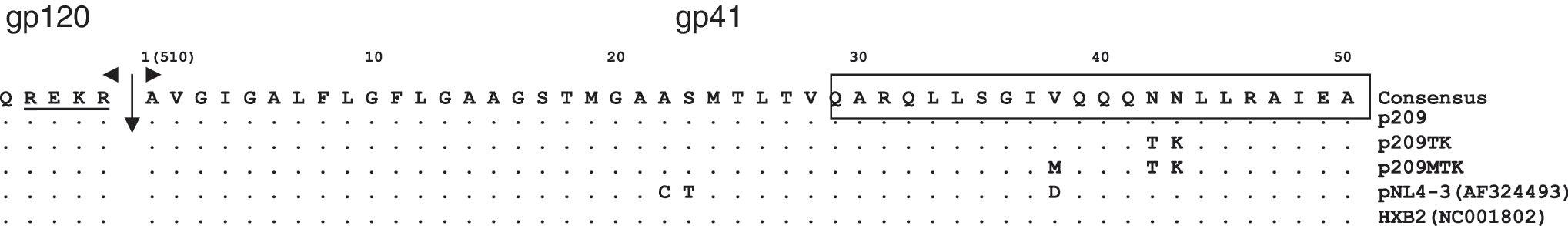
Alignment of the gp120–gp41 cleavage sites and N-terminal regions of the clones with substitutions introduced by site-directed mutagenesis (clone p209). The furin gp120–gp41 cleavage site is marked (↓) and the N-terminal part of HR1 is boxed. The numbering started from the first amino acid (1A) in gp41 which corresponds to A510 in Env (GenBank accession number AF324493).
The HEK293T (ATCC no. CRL11268) and TZM-bl (obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, catalog no. 8129) cell lines were used for the transfection and for the infectivity tests, respectively. The cell cultures were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U of penicillin, 0.1 mg/ml of streptomycin, and
Viruses were generated by transient transfection of the HEK293T cells. For each experiment 5 × 105 cells were transfected with 3 μg of a plasmid and 12 μl of Mirus transfection reagent (Mirus, Madison, WI). The culture supernatants were harvested at 48 h posttransfection and clarified by centrifugation at 3000 rpm (SX 4750 rotor, Allegra X15R centrifuge, Beckman Coulter) for 10 min and at 10,000 rpm for 10 min. Supernatants were filtrated through a 0.45-μm filter (Millipore Corp.) and tested for reverse transcriptase (RT) activity and infectivity. Viruses were isolated from filtrated supernatants by centrifugation through a 20% sucrose cushion at 26,000 rpm (SW 41 rotor, Beckman Coulter) at 4°C for 2 h. Virus pellets were collected in TN buffer and kept at −80°C before use in Western blotting. We further designated viruses without substitutions as “v209” and viruses with substitutions as “v209
The culture supernatants (2.5 μl aliquots) from cells transfected with p209, p209
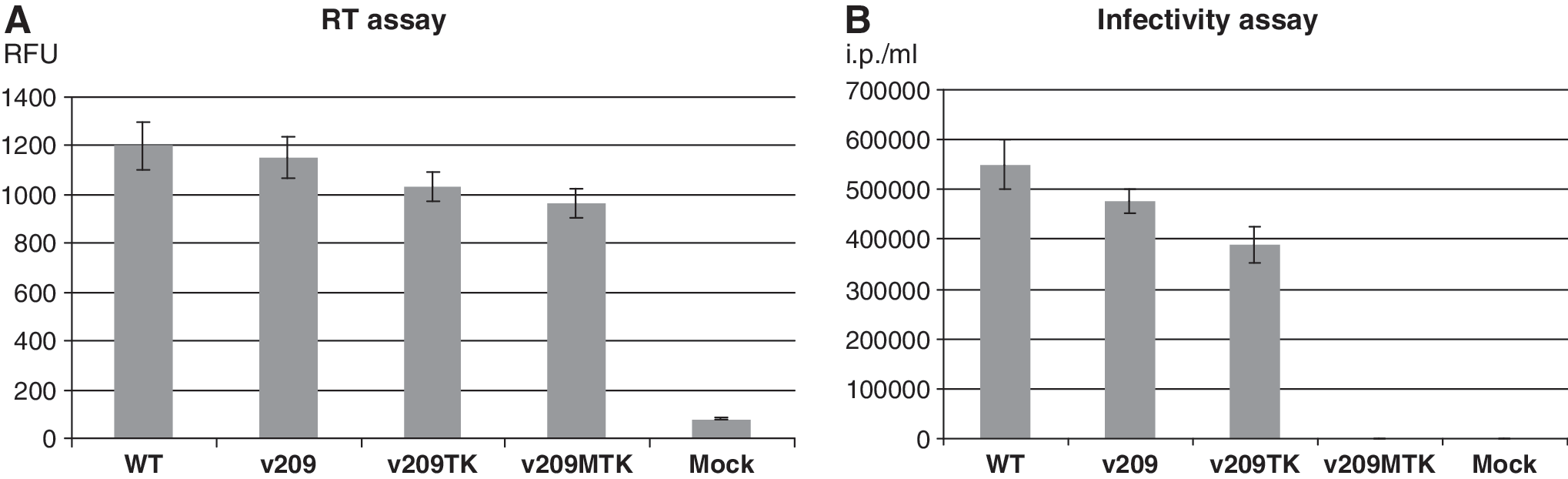
Reverse transcriptase (RT) assay (
Virus infectivity was assessed using the TZM-bl reporter cell line containing cassettes comprising the HIV-1 long terminal repeat (LTR), placed upstream of the lacZ and firefly luciferase genes, respectively. Expression of the reporter cassettes is therefore dependent on the transcriptional activity of the HIV LTR induced by Tat.
Infectivity was estimated in 96-well plates in triplicate using supernatants from two independent transfections. Then 4 × 103 cells per well were plated and incubated overnight at 37°C. The next day when the cell monolayer was about 40–50% confluent, the medium was removed and changed for the serial dilutions of the supernatants from transfected 293T cells in complete medium. The initial dilution contained 20 μl of supernatants from transfected cells and 80 μl of complete medium. To increase the efficacy of infection, the plates were spinoculated at 3000 rpm (SX 4750 rotor, Beckman Coulter Allegra X15R centrifuge) for 35 min at room temperature and then incubated at 37°C. Then 48 h postinfection the cells were washed twice with phosphate-buffered saline (PBS) and fixed with 2% paraformaldehyde for 5 min. Fixed cells were washed with PBS and stained with X-gal (0.5 mg/ml) in PBS containing 5 mM K-Ferric cyanide, 5 mM K-Ferro cyanide, and 2 mM MgCl2. The plates were left in a dark at room temperature for 16 h. The next day cells were washed once with PBS and the blue-stained cells were counted using a light microscope. The wells containing between 5 and 100 blue-stained cells were used for calculation; individual groups of blue-stained cells were counted as single foci of infection
The infectivity test revealed from ∼4 × 105/ml to ∼6 × 105/ml infectious particles in supernatants from HEK293 T cells transfected with p209, p209TK, or pNL4-3 (Fig. 2B). We observed only a minor (∼20–25%) reduction of the infectivity of v209
We next examined by Western blotting whole cell lysates and virions pelleted from the medium of transfected HEK293T cells. Electrophoresis was performed in precast sodium dodecyl sulfate-NuPAGE 4–12% gradient gels (Novex, Invitrogen, San Diego, CA). Proteins were transferred onto BA83 nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany) at 45 V for 2 h. Membranes were blocked with 6% skim milk in PBS with 0.1% Tween 20 (block buffer) for 3 h at room temperature or overnight at 4°C. The membranes were probed with rabbit anti-p24 (HIV-1SF2 p24 antiserum, catalog no. 4250) and goat anti-gp120 (HIV-1 gp120, PB1, catalog no. 36) antibodies. Both sera were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. Human anti-gp41 monoclonal antibodies (mAb) (Chessie 8, epitope PDRPEG) were kindly provided by Dr. George K. Lewis (IHV, UMB). The secondary antibodies were purchased from Zymed Laboratories (Invitrogen Immunodetection). The blocked membranes were incubated in the primary antibodies (1:500) in blocking buffer for 2 h at room temperature. After washing five times (5 min each) in PBS with 0.1% Tween 20 (PBS-Tween) the membranes were incubated for 1 h at room temperature in a corresponding secondary antibodies diluted 1:5000 in blocking buffer. The mAbs were diluted in blocking buffer containing 1% of skimmed milk and the membranes were incubated for 3 h at room temperature, followed by incubation with secondary antibodies as mentioned above. The membranes were washed five times (5 min each) in PBS-Tween, treated for 1 min with Pierce ECL Western blotting substrate (Pierce, Thermo Fisher Scientific Inc., Rockford, IL), and exposed to BioMax Light Chemiluminescence film (Kodak, Sigma-Aldrich).
Western blotting with anti-p24 revealed an efficient cleavage of Pr55gag in the viruses with substitutions (Fig. 3A, B, lower panel, and C). However, the Env processing was remarkably different and uncleaved gPr160 was a predominant protein detected in HEK293T cells transfected with p209
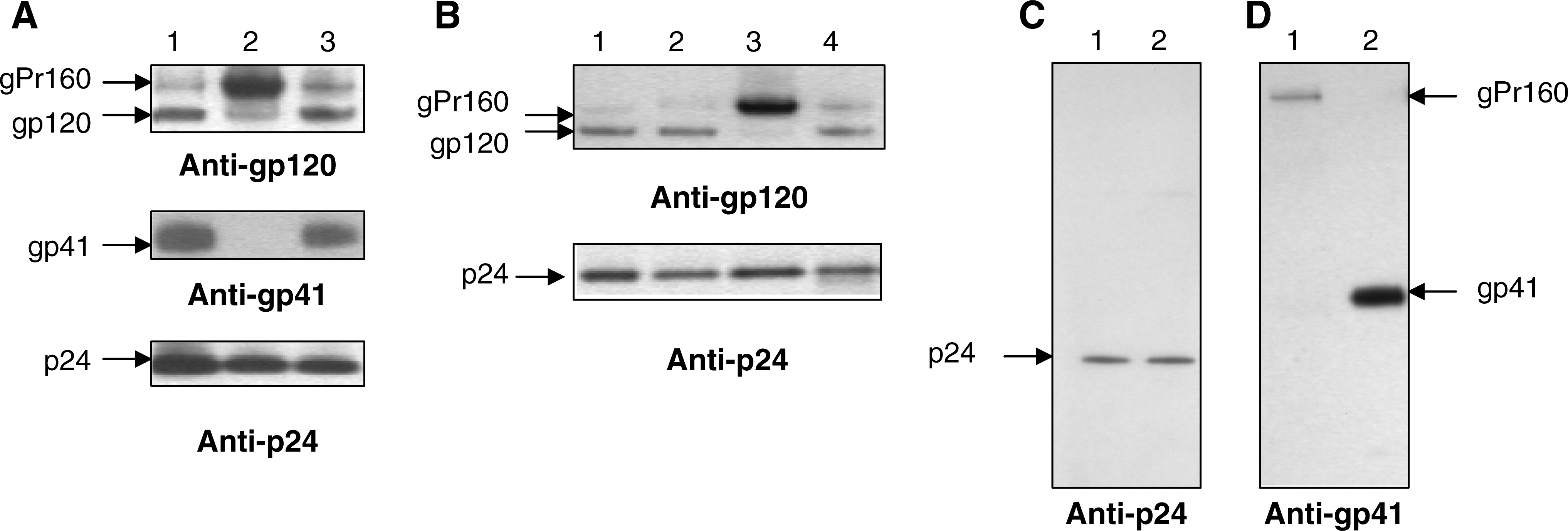
Virus-specific proteins detected by Western blotting in transfected HEK293T cells (
Using the PsiPred program (
The HIV-1 gp41 disulfide loop is conserved among HIV-1 isolates and is a site of noncovalent interaction with gp120. 32 Recently, it has been demonstrated that mutations C598A/C604A (C87A/C93A in TM) in the disulfide loop abolished gPr160 endoproteolytic maturation. 33 Earlier, it was shown that removal of the gp41 glycosylation sites (positions in TM: 100–102, 105–107, and 113–115) disrupted the gPr160 cleavage. 34 Thus, several lines of evidence indicated that certain residues (or combination of residues) in the gp41 ectodomain not in close proximity to the gp120–gp41 cleavage site contributed to the conformation and furin access to the target. Because furin does not have a complex active site and is able to recognize linear peptides containing the target sequence, 35 it seems more likely that the substitutions downstream to the gp120–gp41 cleavage site induce local conformational changes that significantly (if not completely) reduce access of furin to the target sequence. However, it cannot be excluded that more substantial perturbations of the gPr160 conformation took place.
The fact that the V38M + N43T–N44K mutations in viruses from T-20-treated patients have not been reported so far argues against the existence of the compensatory mutations. Nevertheless, remarkable virus plasticity allows speculation that a compensatory mutation(s) may possibly emerge.
We have therefore demonstrated that combined V38M + N43T–N44K mutations disrupt gPr160 cleavage. Furthermore, a minor decline in the RT level and infectivity of the v209
The data provided emphasize the importance of V38 + N43–N44 residues for gPr160 folding, cleavage, and, thus, for virus infectivity. The results obtained may explain why doubly mutated viruses were not detected in AIDS patients under T-20 therapy
Footnotes
Acknowledgments
We thank Dr. Gregory B. Melikiyan and Dr. Claudia Kücherer for their interest and remarks. We are grateful to Dr. Joachim Denner (Robert Koch Institute) for critical reading of the manuscript. The AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH is acknowledged for providing rabbit anti-p24, goat anti-gp120, and TZM-bl cells. This work was supported in part by NIH Grant GM 054787 and by a grant (325-4476-02/3) from the Ministry of Health of Germany.
Disclosure Statement
No competing financial interests exist.
*
This article is dedicated to Professor Alexei I. Morozov (1928–2009).