
Abstract
Human immunodeficiency virus 1 subtype D (HIV-1D) contributes to a significant portion of the HIV-1 disease burden in eastern and central Africa, and is associated with more rapid disease progression. Its viral envelope sequences, particularly in the third variable region (V3), are highly divergent from other major subtypes yet have rarely been studied to date. We evaluated the V3 and select bridging sheet residues of the HIV-1D 94UG114 envelope by alanine-scanning mutagenesis to determine the residues involved in CCR5 usage conservation in the face of sequence variability. We found most single alanine mutations capable of abolishing CCR5 binding, suggesting binding contacts that are highly sensitive to mutation. Despite drastic binding defects across the board, most mutants mediated fusion at or near wild-type levels, demonstrating an ability to accommodate changes in CCR5 affinity while maintaining the ability to complete entry. Three of the alanine mutations did not abolish CCR5 binding but rather resulted in enhanced CCR5 binding. The positions of these residues were found to be conserved between strains of two subtypes, revealing similar V3 elements that suggest a conservation of constraints in V3 loop conformation.
Introduction
H
Clinically, HIV-1D viruses have been associated with increased pathogenicity and more rapid disease progression when compared to other cocirculating subtypes and recombinants. 3 –8 Although it has yet to be determined whether the switch in coreceptor usage from CCR5 to CXCR4 is a cause or consequence of rapid disease progression, it has been noted that subtype D sequences more frequently display properties associated with CXCR4 usage, such as positively charged residues at positions 306 and 320 in the third variable loop (V3) and a higher positive charge in the V3 region overall. 9,10 Phenotypic assays of clinical isolates from Uganda have also revealed a greater likelihood of CXCR4 usage in subtype D viruses compared to subtype A and subtype A/D recombinants. 5,11 As such, although HIV-1D viruses phylogenetically cluster more closely with subtype B viruses, 9,12,13 their envelope sequences are highly divergent relative to other subtypes, being more frequently characterized by length polymorphisms and intraclade sequence variation, especially in the V3 region. 9,14 –16
Although determinants of CXCR4 usage tend to be isolate-specific and distributed through various regions of the envelope, typically in the V1/V2 and V3 regions, 17 –21 work based on subtype B initially identified the V3 as the primary determinant of CCR5 coreceptor usage, and the V3 alone has been shown to confer CCR5 usage to CXCR4-using viruses. 22,23 It is thought that the stem of the V3, along with bridging sheet residues of the C4, interacts with the CCR5 N terminus while the crown of the V3 provides selectivity for CCR5 via its contacts with the CCR5 second extracellular loop (ECL2). Unfortunately, despite a growing body of clinical literature concerning HIV-1D and the unique properties that are likely tied to the env gene, there is a lack of molecular data on the envelope glycoprotein and V3 region of subtype D viruses. Understanding the basis for the observed clinical implications of HIV-1D infection requires examination of how CCR5 use is maintained despite the subtype's characteristically extensive sequence variation. To accomplish this, we undertook scanning alanine mutagenesis in these regions of the infectious molecular clone 94UG114 and analyzed them for CCR5 coreceptor usage through binding and fusion studies.
Materials and Methods
Cells
Cell lines were obtained from the NIH AIDS Research and Reference Reagent Program and maintained at 37°C. Human kidney 293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (DMEM/FBS/PS). U87-CD4-CCR5 cells were cultured in DMEM/FBS/PS with 1 μg/ml puromycin and 300 μg/ml genecitin. Cf2Th/synCCR5 cells 24 were cultured in DMEM/FCS/PS with 3 μg/ml puromycin, 500 μg/ml genecitin, and 500 μg/ml zeocin.
Construction of envelope expression vectors
The HIV-1D molecular clone p94UG114.1.6 was obtained from the NIH AIDS Reagent Program. 25 Alanine-scanning mutagenesis of the V3 region and selected amino acids in the bridging sheet was accomplished by overlap extension polymerase chain reaction (PCR) using primers containing the desired mutations. The DraIII–PpuMI fragment (containing the V3 region) or the PpuMI–SacI fragment (containing the C4 bridging sheet) was swapped into that of TD172, a subcloning vector with the XbaI–XmaI fragment of the p94UG114.1.6 envelope inserted into pTD303-1. The resulting XbaI–XmaI fragment was then inserted back into p94UG114.1.6 to create the full-length envelope mutant. Clones were sequenced to verify the presence of the desired mutations as well as to ensure that no other mutations were introduced during the cloning process.
To generate soluble gp120 (sgp120) expression vectors, we used primers encoding a TAA stop codon at the gp120–gp41 cleavage site. The resulting PCR product was digested with PpuMI and SacI, and inserted into the subcloning vector TD172, and the entire envelope fragment (XbaI–XmaI) was inserted into the pCI vector (Promega) to generate the s94UG mutant panel.
Soluble gp120 binding assay
Five micrograms of DNA from each mutant in the s94UG mutant panel was transfected into 293 cells using the Superfect Transfection Reagent and supernatants were collected after 72 h. The assay for quantifying gp120 binding to CCR5 has been described previously, 26 with some modifications. Cf2Th/synCCR5 cells were plated on a 96-well tissue culture plate at a density of 0.5 × 104 cells and incubated overnight. They were then fixed with 5% formaldehyde for 5 min, and blocked overnight with 5% skim milk in phosphate-buffered saline (PBS). The supernatants were incubated with soluble CD4 (sCD4; Protein Sciences) for 1 hr before being added to the Cf2Th/synCCR5-coated wells. After incubation at room temperature for 1 h, the wells were washed with PBS-Tween 20 and 5 μg/ml of the mouse anti-CD4 antibody M-T441 (Ancell) was added for 1 h. A 1:5000 dilution of a horseradish peroxidase (HRP)-conjugated goat antimouse Ig antibody was subsequently added to the washed wells. One-Step Ultra TMB ELISA (Pierce) was used for color development, and activity was expressed as a percentage of wild-type (WT).
To account for the varying amounts of gp120 in the supernatant, sgp120 was quantified in a sandwich ELISA, using the sheep anti-gp120 polyclonal antibody D7324 as the capture antibody. The gp120 was detected with pooled sera from patients infected with HIV-1 subtype D viruses, followed by incubation with alkaline phosphatase (AP)-conjugated goat antihuman Ig antibody. Envelope gp120 amounts were calculated as a percentage of WT and used to standardize the binding readout using the following formula: % of wild-type binding = [(mutant binding ÷ wild-type binding) × 100] × (mutant expression ÷ wild-type expression).
Soluble CD4 binding assay
Envelope binding to CD4 was studied as previously described. 26 Briefly, 2 μg/ml of D7324 was used to coat 96-well plates overnight, and sgp120 from 293 cells transfected with the envelope expression vectors was incubated in each well, followed by sCD4. After washing off excess sCD4, bound CD4 was detected with 5 μg/ml M-T441. A 1:5000 dilution of HRP-conjugated goat antimouse Ig antibody was added to the washed wells and incubated for 1 h. Color development with One-Step Ultra TMB ELISA was stopped with 1 M sulfuric acid, and signal was quantified at OD450.
Cell–cell fusion
The cell–cell fusion assay was performed as previously described, 26 with some modifications. In all, 293 cells were plated in 96-well tissue culture plates at a density of 8 × 104 cells for transfection with 0.5 μg of DNA. After 48 h, the cells were infected with the recombinant vaccinia vTF7-3 (NIH AIDS Research and Reference Reagent Program) at a multiplicity of infection (MOI) of 10. 27 This construct expresses T7 RNA polymerase under the control of the early/late vaccinia promoter. An equivalent number of U87-CD4-CCR5 cells was infected with 10 MOI of recombinant vaccinia vCB21R-lacZ (NIH AIDS Research and Reference Reagent Program), which contains the lacZ gene under the control of the T7 promoter. The cells were washed in PBS after 1.5 h and left to incubate overnight. The U87-CD4-CCR5 cells were detached by scraping and then mixed with the 293 cells in the presence of 40 μg/ml AraC. After a 3-h incubation, the cells were washed and lysed with Reporter Lysis Buffer (Promega) for 10 min at room temperature. Fusion was then quantified using the β-Gal Assay Kit (Invitrogen) to assay for β-galactosidase activity according to the manufacturer's instructions, and absorbance was read at 405 nm.
Radiolabeled immunoprecipitation assay
For metabolic labeling, [35S]cysteine in cysteine-free,
Results
Construction of p94UG114.1.6 mutant panels
The HIV-1 subtype D molecular clone p94UG114.1.6, an infectious subtype D reference strain, 12 was selected for alanine-scanning mutagenesis due to its use of CCR5 as the main chemokine coreceptor for viral entry. 25 The V3 region of p94UG114.1.6 env consists of 34 amino acid residues (Fig. 1A). The 32 amino acids within the V3 loop, excluding the terminal cysteine residues involved in disulfide bond formation, and five residues in the C4 region were singly mutated to examine the effect of these amino acids on envelope expression. Most residues were mutated to alanine, except for the two naturally occurring alanines that were mutated to glycine. To generate these desired mutations, fragments generated from overlap extension PCR were inserted into a TD172 subcloning cassette containing the 94UG114.1.6 XbaI–XmaI env sequence; this XbaI-XmaI fragment was subsequently excised and inserted into p94UG114.1.6 to generate the mutant molecular clones (Fig. 1B). The s94UG sgp120 expression vectors were prepared by generating a stop codon at the gp120–gp41 cleavage site in the TD172 subcloning mutant panel and inserting the envelope containing the premature stop codon into the pCI mammalian expression vector.
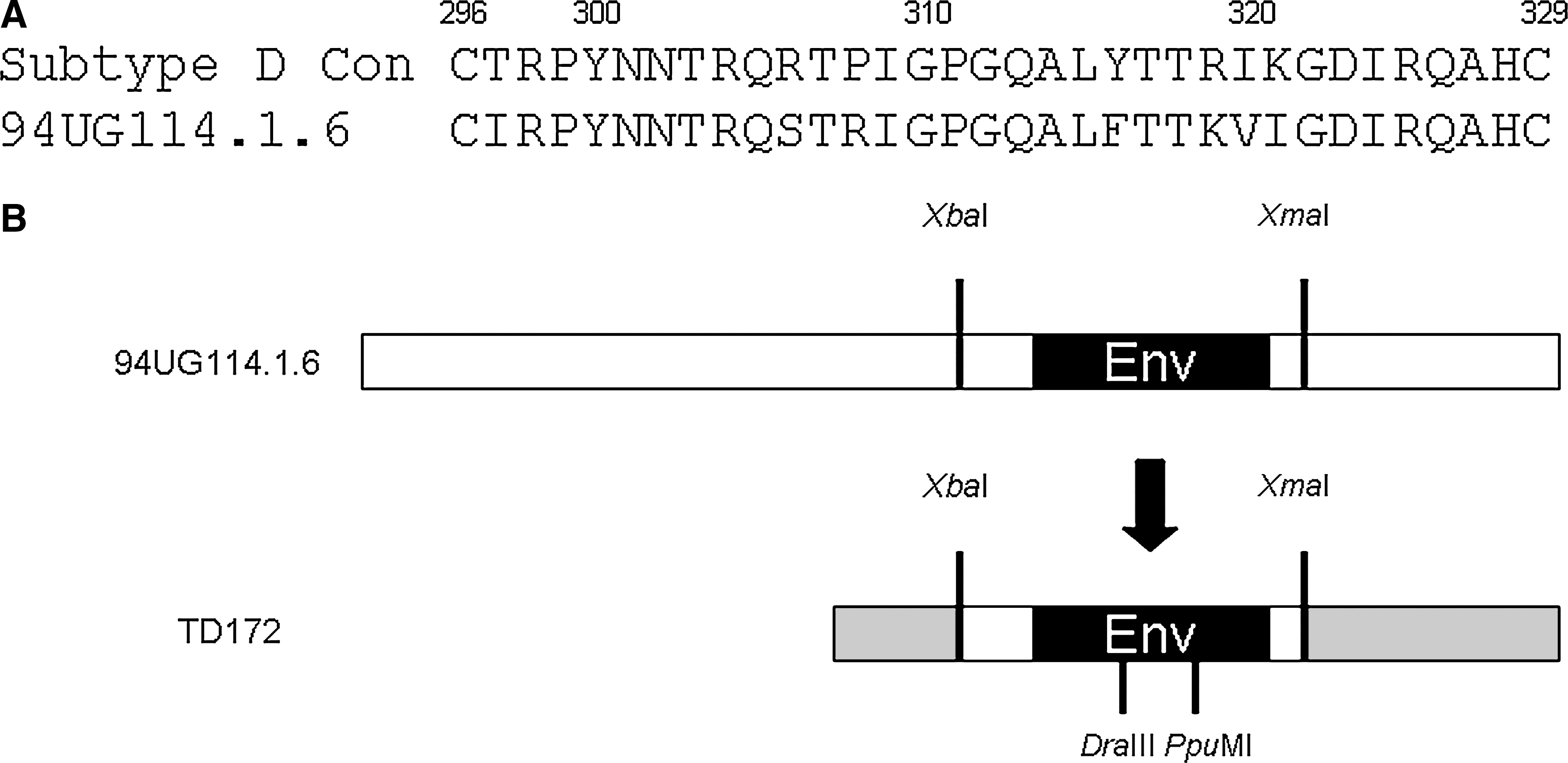
HIV-1 subtype D 94UG114.1.6 molecular clone. (
Ability of envelope mutants to mediate CCR5 binding
To evaluate the effect of these V3 and select C4 mutations on CCR5 coreceptor binding, sgp120 was harvested from the supernatant of 293 cells transfected with the s94UG mutant panel. The relative amounts of sgp120 were measured by ELISA using the sheep anti-gp120 D7324 to capture the sgp120, and the results were calculated as a percentage of WT expression. To evaluate CCR5 binding, sgp120 was precomplexed with sCD4 and assayed by ELISA for binding to CCR5 on the surface of Cf2Th/synCCR5 cells. Coreceptor binding was quantified as a percentage of WT standardized by envelope expression (Fig. 2A). We found that the 94UG114 virus poorly accommodated mutations in regions associated with CCR5 coreceptor binding, with most single mutations (29 of 37) reducing CCR5 binding to under 50% of WT activity and the majority of those (24 of 29) to under 25% of WT activity. We observed that reduced binding to CCR5 resulted from mutations distributed across both the stem and crown regions of the V3.
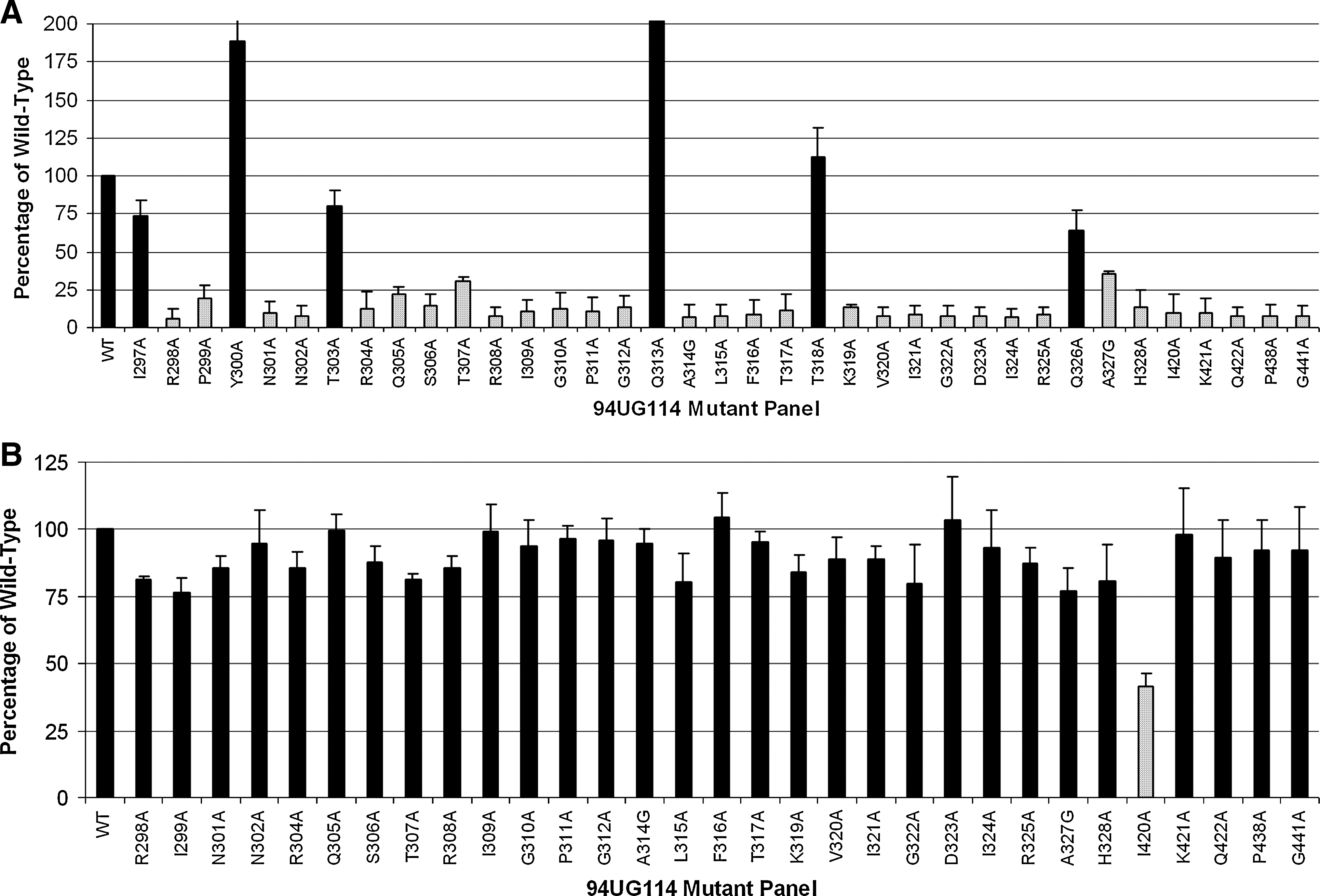
Receptor binding by the 94UG114 mutant panel. (
Only 6 of the 37 mutants exhibited activity above 50% of WT (I297A, Y300A, T303A, Q313A, T318A, and Q326A). Of these, half (Y300A, Q313A, and T318A) had enhanced binding to CCR5, with activity >100% that of WT.
Ability of envelope mutants to bind CD4
To verify that the observed defects in CCR5 binding were not due to the effect of those mutations on the ability of gp120 to bind the CD4 receptor, the mutants were assayed for CD4 binding capacity. As shown in Fig. 2B, only I420A showed a marked decrease in CD4 binding, with less than 50% of WT activity. This role of I420 in CD4 binding has been described previously with subtypes B and C. 26,28 Other than at this position, the introduced mutations did not notably affect the ability of mutant envelopes to bind to the CD4 receptor, and the effects of these single mutations in the V3 and C4 on CCR5 binding are not due to defects in CD4 binding.
Ability of envelope mutants to mediate cell–cell fusion
Studies have shown a distinct difference between coreceptor binding as demonstrated by an sgp120 binding assay versus a cell–cell fusion assay. 26,29 –31 We have previously demonstrated that this discrepancy is not necessarily introduced by assay differences, but that a given envelope may have distinct capacities for CCR5 binding and membrane fusion. 32 Having observed the drastic loss of CCR5 binding associated with the majority of single mutations introduced into the V3 region, we proceeded to investigate whether membrane fusion could still occur despite this decrease in binding capacity.
To investigate the fusion activity mediated by native envelope, we used a reporter gene activation assay to measure the ability of the V3 and C4 mutants to mediate cell–cell fusion. The extent of fusion, as shown by β-galactosidase activity, is expressed as a percentage relative to that of the WT virus, which was set at 100% (Fig. 3). Two mutations (R298A, A327G) in the V3 loop strongly affected the ability of the envelope to mediate fusion, resulting in <50% of WT β-galactosidase activity. Another five mutations (N301A, N302A, T303A, F316A, and T317A) yielded clones exhibiting around 50% of WT activity. Five of these residues are located in the stem region and two (F316A and T317A) are in the periphery of the crown.

CCR5 coreceptor use by the 94UG114 mutant panel. In all, 293 cells expressing surface gp120 were mixed with U87-CD4-CCR5 cells, and fusion was quantified by β-galactosidase activity.
Among the C4 bridging sheet mutants, I420A and P438A exhibited decreased fusion activity, while two of the three remaining mutants had around 50% WT activity. It has been observed previously, however, that the I420A mutation adversely affects CD4 binding, 26,28 and it is likely that this defect is reflected in downstream functions.
While the mutants exhibiting fusion defects (Fig. 3; R298A, N301A, F316A, T317A, A327G, I420A, and P438A) are also associated with poor binding, it is likely that the severe defect in CCR5 binding may not be the primary reason. This is due to the observation that most mutants with severely deficient binding are capable of mediating WT or near-WT levels of fusion activity. The three mutants associated with enhanced binding (Y300A, Q313A, and T318A) did not mediate noticeably higher levels of fusion either.
V3 mutations alter envelope expression and processing
To determine whether there were changes in envelope expression that may have affected fusion readout among these particular mutants, transfected 293 cells were incubated with [35S]cysteine overnight prior to collection of cell lysate and supernatant, and viral proteins were immunoprecipitated using pooled sera from HIV-1D-infected patients. Analysis by SDS–PAGE revealed differential cellular and supernatant expression of gp120 compared to WT (Fig. 4). Quantification of relative gp120 levels in each fraction by densitometric analyses (Table 1) revealed that these substitution mutants were characterized by decreased amounts of gp120 in the cell lysate and increased amounts of sgp120 in the culture supernatant. The very low levels of A327G gp120 in the cell lysate did not correspond to an accordingly large increase in supernatant gp120 levels, suggesting that this mutation may have an impact on other envelope properties. Changes associated with the N301A envelope were not as drastic as those seen with other mutants with poor fusion. The impact of this mutation may be due more to an effect of the alanine substitution on the ability of the envelope to mediate fusion. Overall, single mutations in the V3 appear to affect fusion and entry function by altering gp120 association with gp41 and reducing the availability of gp120 on the cell surface.
Envelope expression by select 94UG114 mutants. (
Discussion
To address the gap in our understanding of HIV-1D viruses and their molecular properties, we analyzed the envelope properties of an HIV-1D virus by examining the determinants of CCR5 coreceptor usage in the V3 and C4 regions of gp120. For this purpose, we selected the p94UG114.1.6 isolate, a subtype D reference strain available for study as an infectious, full-length molecular clone. Alanine scanning mutagenesis was performed on residues in the V3 and select bridging sheet residues of the C4 associated with CCR5 binding 33 in order to study the role of the mutated residues on envelope-mediated entry. In particular, we were interested in investigating the extent of differences and similarities between the subtype D virus and subtypes B and C viruses in their capacity to accommodate mutations that affect coreceptor binding and to maintain CCR5 usage.
We found that single alanine mutations had little impact on fusion for most residues in the V3. The R298A mutation significantly affected both fusion and binding. This provides another strong support for the importance of this residue for V3 activity, which has been observed in several previous studies to result in decreased CCR5 usage and infectivity on substitution of this highly conserved residue in subtype B (HXB2-JRFL) and subtype C (HXB2-1471) envelopes. 26,28,34 Mutation of the three residues spanning an N-linked glycosylation site in the N-terminus of the V3 loop (N301A, N302A, and T303A) reduced fusion levels by approximately 50%. The role of the N301A mutation in fusion, like the R298A mutation, was conserved across isolates from subtypes B and C, with similar detrimental effect. 26,28 It is unlikely to be due to the loss of N-linked glycosylation, however, as N302A also reduced fusion by similar amounts for both 94UG114 and HXB2-1471. The three-residue stretch flanking the V3 crown (L315A, F316A, and T317A) also displayed reduced fusion activity compared to neighboring residues, with the latter two under 50% WT activity. This involvement of the crown region is distinct from that seen with subtypes B and C, and may be due in part to the changes in cellular expression of gp120.
The decreases in fusion capacity may not be directly associated with CCR5 binding, as most mutants were characterized by a drastic loss of CCR5 binding yet were able to mediate cell–cell fusion. Previous work with subtype B suggested that such discrepancies between fusion and binding data are not necessarily due to assay differences. 32
Unlike previous studies examining subtype B coreceptor binding by similar methods, 28,32 however, the number of single amino acid mutants found to be critical in mediating CCR5 interactions is much higher. The V3 of 94UG114 appears to be extremely sensitive to mutations with respect to coreceptor binding compared to what has been observed with JRFL, with 83.4% (31 of 37 mutants) exhibiting <50% of WT activity, and 93.5% of those (29 of 31 mutants) having <25% activity. In comparison, 65.8% (25 of 38) of HXB2-JRFL V3 alanine-scanning mutants had less than 50% of WT activity, and only 52% of these (13 of 25 mutants) had under 25% activity. 26 Among these, mutated V3 crown residues tended to retain binding activity, although this trend was not observed with subtype D mutants.
The detection of defects in coreceptor binding is a standard means of studying the envelope and its interaction with CCR5. However, little attention has been paid to mutations observed to enhance coreceptor binding. Previous studies using the HXB2-JRFL subtype B chimera pinpointed a number of specific alanine mutations in the V3 capable of enhancing binding: T297A, N300A, A314G, T317A, E320A, and Q327A. 26,32,35 Although these observations cannot be generalized across the gamut of HIV-1 isolates from such data alone, with our analysis of the 94UG114 envelope, we report three residues in the V3 conferring enhanced CCR5 binding, namely Y300A, Q313A, and T318A. Of these, the role of the residue at position 300 is conserved between the two strains, whereas positions 313 and 318 involve a single amino acid shift. This shift may be due to the one amino acid difference in V3 length between JRFL and 94UG114. Furthermore, although most alanine mutations in the 94UG114 V3 resulted in drastic loss of CCR5 binding, I297A and Q326A, which correspond to the JRFL T297A and Q327A mutants, are among the very few mutants (6 of 37) retaining any significant CCR5 binding activity. It is possible that a V3 less sensitive to mutation may display enhanced binding for these mutants as well.
Within subtype D isolates, residues at positions associated with CCR5 binding enhancement (Y300, Q313, and T318) are moderately conserved. In a 1998 amino acid frequency analysis using 133 HIV-1D isolates, 36 alanine polymorphisms were seen only in 1% of viruses at position 300, none at 313, and in 2% at 318. Similarly, alanine is rarely seen at positions 297, 303, and 326, with frequencies of 5% at 297 and under 1% at 303 and 326. This suggests that despite the enhanced CCR5 binding capacity of mutants carrying alanine at one of these three positions, there does not appear to be positive selection for these alanine variants.
HIV-1B JRFL positions associated with enhanced CCR5 binding likewise rarely have alanine. Only at position 317 is alanine a common polymorphism, with threonine seen at the position in 56% of isolates and alanine in 41%. This property is shared with subtype D viruses but not with other subtypes, which have a marked preference for alanine at 317. 37 The A/T polymorphism at this position has been described as conferring context-dependent differential susceptibility to entry inhibitors PSC-RANTES, enfuvirtide, and TAK-779. 37 Our data suggest the possibility that these polymorphisms may affect inhibitor sensitivity through modulation of CCR5 binding affinity. As these alanine mutations (and one alanine-to-glycine mutation) involve a decrease in size of the side chains at their respective positions, changes in CCR5 binding may be due to increased flexibility of the local V3 structure or increased accessibility due to decreased steric hindrance. It is interesting to note that increased cell–cell fusion is not associated with enhanced binding for any of these mutants. The lack of an observed gain in this downstream function may explain why neither 94UG114 nor JRFL takes advantage of this enhanced binding capacity.
The identification of cross-clade conservation of positions capable of conferring enhanced coreceptor binding despite sequence divergence as well as structural and functional vulnerability to mutations provides new insight into the molecular anatomy of the V3 loop. These strong parallels pinpoint constraints in the V3 loop that may be associated with convergence in coreceptor usage. The observation that enhanced binding capacity conferred by the alanine substitutions was not selected for in the isolates studied raises intriguing questions about the role of the V3 loop in viral adaptation and fitness.
Footnotes
Acknowledgments
We would like to thank Mary Fran McLane for preparation of vaccinia stocks and other assistance and Lendsey Melton for editing the manuscript. S.T. was supported by Grant NIH D43 TW00004 from the Fogarty International Center.
Author Disclosure Statement
No competing financial interests exist.