
Abstract
With considerable capacity for genetic diversification, new HIV-1 genotypes have been reported over the years. Three HIV-1 isolates previously genotyped as B using gag and env sequences were completely sequenced and reanalyzed. Several amino acid mutations were found in vif, rev, and nef genes but not in gag or env sequences. These alterations have not previously been reported in Hong Kong. The investigation of phylogenetic relatedness revealed that a region of the vif of the studied Hong Kong isolates subtype B cluster contains several subtype D signature amino acid residues. Several unique mutations on vif in these three isolates were also identified.
H
Serum samples of Hong Kong patients were obtained from the Centre for Health Protection, Department of Health, Hong Kong. These samples were collected during the period from January 2002 to December 2006 with demographic information available. The viral load of the samples ranged from 2130 to 2,200,000. The entire HIV genome was amplified with nested polymerase chain reaction (PCR) using degenerated primers designed with reference to the HIV-1 HXB2 reference (primer sequence available on request). Nested PCR was performed with Taq polymerase (Genesys limited) and Takara Ex Taq polymerase (Takara). Both rounds of PCR were subjected to 5 min denaturation at 94°C, followed by 10 cycles of 94°C for 30 s, 45°–57°C for 30 s, and 68°C for 1–3 min, then 30 cycles of 94°C for 30 s, 43°–55°C (2°C lower than the first 10 cycles) for 30 s, 68°C for 1–3 min, and a final extension at 68°C for 10 min. PCR product was purified with 3 U shrimp alkaline phosphatase (USB) and 30 U exonuclease I (USB) to remove the leftover primers and dNTPs. PCR products was directly sequenced using sense and antisense degenerated primers and fluorescently labeled dideoxy chain terminators (BigDye Terminator v3.1 Cycle Sequencing Assay, Applied Biosystems) in ABI Prism 3100 Genetic Analyzer (Applied Biosystems). Sequence contig assembly and initial analysis were performed using the Seqman module of DNASTAR Lasergene 7.0 (DNASTAR).
The full-length sequences of three of the isolates of a subtype B cluster were verified and deposited in the NCBI GenBank database with accession numbers of FJ460499, FJ460500, and FJ460501. The viral genomes of the three isolates were about 9 kb in size spanning the gag gene to the nef gene. Basically, all the isolates were genotyped as subtype B using the NCBI Genotyping Tool (
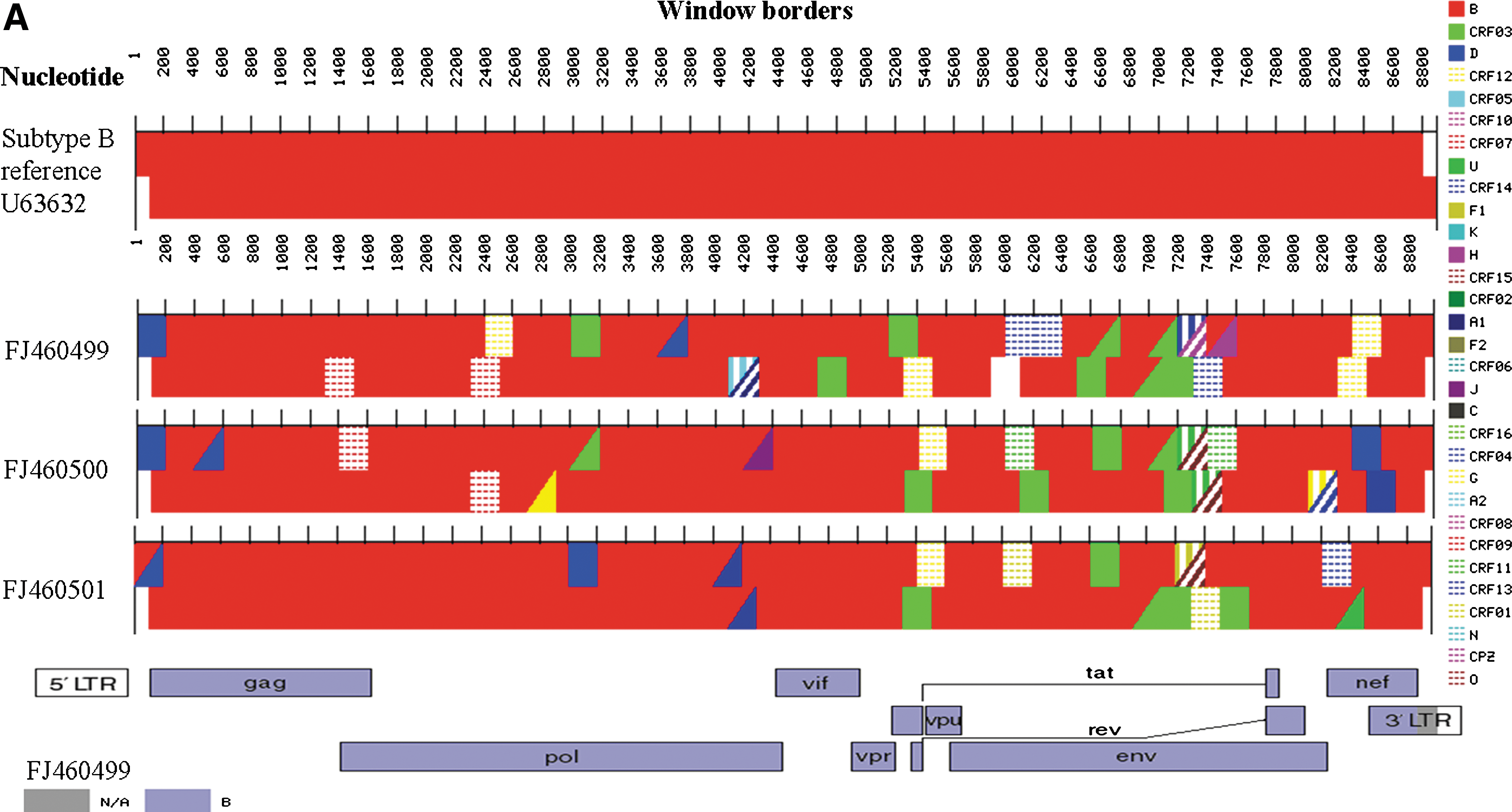
(


Whereas genotyping using our full-length sequences did not affect previous genotyping results using gag and env sequences, we were able to identify many amino acid mutations using the complete genome sequences. Alignment with subtype B reference sequences retrieved from the NCBI Genotyping Tool database of 2004 and 2005 using BioEdit version 7.0.9.0 revealed several amino acid mutations in vif, rev, and nef proteins (Fig. 1B). 9 A similar mutation pattern was not noticed in the gag and env proteins, which were usually used for genotyping. The highly altered sites of these three genes were then searched against NCBI protein databases using the protein-BLAST algorithm (Table 1). Both rev 8–32 (ACJ76659 aa 8–32) and nef 8–57 (ACJ76662 aa 8–57) were aligned to subtype B reference sequences only. Only the vif 93–141 (ACJ76656 aa 93–141) sequences are nearly equidistant from subtypes B and D. Such an alignment was masked if the full-length vif protein sequence (ACJ76656 aa 1–193) was included in the protein-BLAST, in which case only subtype B sequences could be identified.
The vif gene has been shown to be a “hot spot” for genome recombination. 10,11 It has also been used to determine the evolutionary pattern of HIV-1. 12 Thus, we compared the amino acid sequences on vif between our isolates and subtype D isolates as identified by the protein-BLAST. Several amino acid alterations in our isolates, when compared to subtype B references, were also found in the vif proteins of subtype D (Fig. 1C). We have identified four alterations that were exclusively confined to our isolates. They are vif93G, vif105K, vif120V, and vif141Q (Fig. 1C). To further confirm the association with subtype D, multiple alignments of sample and reference vif sequences were performed for phylogenetic analysis using the neighbor-joining method provided in BioEdit. Results showed that the three Hong Kong isolates were in a group distinct from subtype B or D (Fig. 1D).
There are several functionally conserved domains in the vif protein. In maternal–fetal transmission of HIV, vif protein was conserved at cysteine 114, cysteine 133, and serine 144. 13 In addition, vif K22, RH41/42, DR14/15, and W79 were found as crucial residues for interaction with host APOBEC3 and therefore suppression of host antiviral proteins. 14 Interestingly, lysine 22 (K22), a key residue on vif for degrading the host antiviral protein A3G was mutated in our reported isolates but not in other reference sequences of subtype B or D (Fig. 2). However, the functional significance of this sequence variation remains to be confirmed.

The vif K22, one of the functional residues of vif in the degradation of the host antiviral protein A3G, was mutated in the reported sequences.
In conclusion, in this study we have shown the importance of whole genome sequencing in identifying unique mutations in an emerging HIV-1 cluster of subtype B in Hong Kong.
Sequence Data
Nucleotide sequences of the three Hong Kong HIV-1 isolates were deposited in NCBI GenBank with accession numbers of FJ460499, FJ460500, and FJ460501.
Footnotes
Acknowledgment
This project team is supported by the AIDS Trust Fund of the Hong Kong SAR Government.
Disclosure Statement
No competing financial interests exist.