
Abstract
Late diagnosis of HIV-1 infection is quite frequent in Western countries. Very few randomized clinical trials to determine the best antiretroviral treatment in patients with advanced HIV-1 infection have been performed. To compare immune reconstitution in two groups of very immunosuppressed (less than 100 CD4+ cells/μl), antiretroviral-naive HIV-1-infected adults, 65 patients were randomly assigned in a 1:1 ratio to receive zidovudine + lamivudine + efavirenz (group A, 34 patients) or zidovudine + lamivudine + ritonavir-boosted indinavir (group B, 31 patients). The median (interquartile range) CD4+ cell increase after 12 and 36 months was +199 (101, 258) and +299 (170, 464) cells/μl in the efavirenz arm and +136 (57, 235) and +228 (119, 465) cells/μl in the ritonavir-boosted indinavir arm (p > 0.05 for all time points). The proportion (95% confidence interval) of patients achieving HIV-1 RNA levels under 50 copies/ml was significantly greater in the efavirenz arm at 3 years by the intention-to-treat analysis [59% (41%, 75%) vs. 23% (10%, 41%)], whereas no differences were found in the on-treatment analysis. Immune activation (CD8+CD38+ and CD8+CD38DR+ T cells) was significantly lower for the efavirenz arm from month 6 to month 24. Adverse events were more frequent in the ritonavir-boosted indinavir arm. Almost all cases of disease progression and death were observed in the first year of treatment, with no significant differences between the two arms (p = 0.79 by the log-rank test). At 1 and 3 years, the immune reconstitution induced by an efavirenz-based regimen in very immunosuppressed patients was at least as potent as that induced by a ritonavir-boosted protease inhibitor-based antiretroviral regimen.
Introduction
L
Highly active antiretroviral therapy (HAART) has been shown to result in good control of HIV-1 infection when started in HIV-1-infected individuals with a CD4+ T lymphocyte count above 200 cells/μl. 5 Current recommendations support initiating antiretroviral therapy with two nucleoside reverse transcriptase inhibitors (NRTIs) plus a nonnucleoside reverse transcriptase inhibitor (NNRTI) or a pharmacologically enhanced protease inhibitor (PI), that is, a boosted PI. 6 However, a late diagnosis of HIV-1 infection means that some patients must start HAART when HIV-1 has already severely impaired their immune function. Furthermore, international guidelines do not specify which is the best antiretroviral treatment in patients with a low CD4+ cell count, since clinical trials on HAART usually exclude patients with less than 100 cells/μl and recent opportunistic infections (OI). In this clinical setting, physicians tend to prefer the boosted PI-based antiretroviral regimens, 7 probably because of their stronger genetic barrier, thus avoiding the selection of resistant viral strains that could affect long-term virological response. 8 However, very few controlled studies have directly compared the immunological, virological, and clinical outcomes of an NNRTI-based antiretroviral regimen with those of a boosted PI-based antiretroviral regimen in very immunosuppressed patients. 9,10
The aim of this randomized clinical trial was to compare the immune reconstitution after NNRTI (efavirenz)-based HAART with that after a boosted PI (indinavir-ritonavir)-based regimen in severely immunosuppressed antiretroviral-naive HIV-1-infected patients.
Materials and Methods
Design overview
In this open-label trial, HIV-1-infected patients with a CD4+ cell count lower than 100 cells/μl were randomly assigned to receive an NNRTI-based antiretroviral regimen or a boosted PI-based regimen. Enrollment was between November 2001 and January 2003 and the last patient visit was on July 7, 2006. The study was approved by the ethics committees of the participating hospitals and the Spanish Medicines Agency, and conducted in compliance with the Declaration of Helsinki, good clinical practice guidelines, and local regulations. Patients were adequately informed and signed a written informed consent form before enrollment.
Setting and participants
This trial was conducted in six infectious diseases clinics in Spain. Study investigators recruited participants from among their patients after reviewing their medical records and completing screening procedures to assess eligibility. HIV-1-infected adults (age over 18 years) were eligible if they were antiretroviral naive and had a baseline CD4+ cell count below 100 cells/μl. Patients were excluded if they presented an active opportunistic disease requiring parenteral treatment or any contraindication to the study drugs. Female patients were excluded if they were pregnant or breastfeeding.
Randomization and interventions
A centralized computer-generated process assigned eligible patients to one of two treatment arms. Eligible patients were randomly assigned in a 1:1 ratio to receive zidovudine (ZDV, 300 mg bid) plus lamivudine (3TC, 150 mg bid) (Combivir) with either efavirenz (Sustiva 600 mg qd) as the NNRTI or indinavir-ritonavir (Crixivan/Norvir 800/100 mg bid) as the ritonavir-boosted protease inhibitor-based regimen. Patients with previous clinical events included in category C of the 1993 classification of the Centers for Disease Control and Prevention 11 started antiretroviral treatment between 2 and 6 weeks after diagnosis.
Outcomes and follow-up
Screening included a clinical assessment and laboratory evaluations: plasma HIV-1 RNA, T-lymphocyte subsets, hematology, and clinical biochemistry. After randomization, on-study evaluation included clinical visits at baseline and weeks 2, 4, 8, 12, 24, 36, 48, and every 3 months thereafter. Plasma HIV-1 RNA was assessed at each visit using the local HIV-1 assay. Qualitative immunological studies were performed at baseline and every 6 months until month 24. Viral resistance at the time of virological failure was tested for all patients who met the criteria for virological failure. Adherence to therapy and changes in body fat distribution were not measured.
Outcomes and measurements
This study compared the median increase in the number of CD4+ lymphocytes at 12 months in the two arms as its primary objective, with a secondary comparison at months 24 and 36. Secondary endpoints were the proportion of patients with a plasma HIV-1 viral load below 50 copies/ml, the incidence of side effects, qualitative analysis of immune reconstitution, disease progression, and death. Virological failure was defined as detectable HIV-1 RNA levels (at least 50 copies/ml) in two consecutive determinations. Treatment failure was defined as virological failure, loss to follow-up, treatment arm switch or discontinuation of study medication because of toxicity or for any other reason (changes within the same class of antiretrovirals were allowed), clinical disease progression (any new clinical event included in category C of the 1993 CDC classification 11 ), or death.
Immunological studies
Peripheral blood mononuclear cells (PBMCs) were obtained by separation on a Ficoll-Hypaque (Biomedics-Biomérieux, Madrid, Spain) centrifugation gradient. Subpopulations of CD3+, CD4+, and CD8+ cells were determined by three-color flow cytometry at baseline and at months 6, 12, and 24 thereafter. The following monoclonal antibodies were used: CD3-peridinin chlorophyll protein (PerCP), CD4-PerCP, CD8-PerCp, CD4-fluoroisothiocyanate (FITC), CD8-phycoerythrin (PE), HLA DR-FITC, CD28-PE, CD38-PE, CD45RO-PE, and CD45RA-FITC (all from Becton Dickinson, Mountain View, CA). Mouse immunoglobulin isotypes conjugated with Per-CP, PE, or FITC were always used as negative controls for nonspecific binding. The stained cells were analyzed on a FACSCalibur (Becton Dickinson, San Jose, CA) flow cytometer. Lymphocytes were gated on the basis of forward and side scatter parameters. The gating region was referred to an FL3/SS histogram, where an FL3+(CD3+, CD4+, or CD8+) region was defined. This region was further analyzed for the expression of FL1 and FL2. Data were analyzed using CELLQUEST software (Becton Dickinson).
PBMC proliferation assays were performed as described elsewhere. 12 PBMCs were washed twice and resuspended at 2 × 106/ml in protein-free medium X-VIVO 10. Cultures were performed in triplicate in 4- or 7-day assays using 105 or 2 × 105 cells/well in 96 round-bottomed microplates. Cells were cultured in the absence or presence of PHA 0.5%, OKT3 10 ng/ml alone or in combination with anti-CD28 100 μg/ml, PWM 10 μg/ml, tetanus toxoid 2750 U, cytomegalovirus antigen 10 μg/ml, and recombinant HIV-1 proteins (p24 and gp160) 5 μg/ml. Incorporation of tritium-labeled thymidine was assessed for the last 18 h of culture. Results are expressed as the stimulation index calculated for each sample as the ratio between the counts per minute (cpm) for the stimulated cultures and the cpm for the nonstimulated cultures. Positive antigen-specific responses were defined as more than 3000 cpm and a stimulation index greater than 3.
Virological studies and resistance tests
Plasma HIV-1 RNA copy levels were evaluated using the commercially available AMPLICOR quantitative restriction transcriptase-polymerase chain reaction assay (Roche Diagnostic Systems, Branchburg, NJ) according to the manufacturer's instructions. The lower limit of detection of the assay was 200 copies/ml. Samples with less than 200 copies/ml were retested using Ultra Direct Assay (Roche Molecular Systems, Alameda, CA) with a lower limit of quantification of 50 copies/ml.
In cases of virological failure, serum samples were obtained and stored at −80°C until genotypic resistance tests were performed. All samples were tested using the ViroSeq HIV-1 genotyping system according to the manufacturer's instructions (Applied Biosystems, Foster City, CA).
Adverse events
Evaluation of adverse events included spontaneous patient reports, open-ended questioning by the physician, physical examination, and assessment of laboratory test abnormalities. These were reported on case report forms in which the investigator was asked to assess the possible cause and the relationship with the study drugs. The severity of toxic effects was assessed using the AIDS Clinical Trial Group toxicity grading scale. 13
Statistical analysis
Patients were followed for the entire trial regardless of whether they prematurely discontinued the assigned therapy. All randomized patients, except those who were found to have violated an entry criterion and those who never started the study medication, were included in the analysis. In the intention-to-treat analysis, treatment was considered to have failed in all patients who died, had a new category C event, withdrew consent, were lost to follow-up, had virological failure, or changed the antiretroviral drug class because of an adverse event, but not if the new drug was from the same class (from efavirenz to nevirapine or from indinavir/ritonavir to lopinavir/ritonavir or atazanavir/ritonavir). In the analysis of patients by treatment received (on-treatment analysis), treatment failure was defined by death, the appearance of a new category C event, or virologic failure. Data on patients who withdrew consent, were lost to follow-up, changed treatment arm, or stopped study medication were censored.
The sample size was calculated on the basis of the immunological endpoint and the immune response was based on a study that overviewed the effectiveness of triple combination therapies in antiretroviral-naive patients who did not have advanced infection 14 and in a retrospective case-control study in patients who did. 15 The sample size was computed to detect differences of 50 cells/μl in the CD4+ lymphocytes at week 48, assuming a mean increase of between 168 cells/μl (95% CI, 145–191) 14 and 185 cells/μl 15 in the protease inhibitor arm. A total of 30 patients per group were required for the noninferiority assessment with a two-sided alpha level of 0.05 and a statistical power of 80%.
Continuous variables were described as the median with the interquartile range and were represented with a box-whiskers plot. Categorical variables were represented as an absolute value and percentage of the total group. Fisher's exact test was used to compare dichotomous variables. Differences in continuous variables between the groups were analyzed using the Mann–Whitney test. The time to disease progression and death was estimated with the Kaplan–Meier method. The equality of the distributions of the times to an event among the groups was estimated using the generalized log-rank test.
Statistical analysis was performed using Stata software (StataCorp 2003, Stata Statistical Software, Release 8.0, College Station, TX).
Results
Baseline patient characteristics
Between November 2001 and January 2003, 76 patients were assessed for inclusion in the study. Of these, 70 were found eligible and were randomized to the treatment arms; four patients in the indinavir/ritonavir arm and one patient in the efavirenz arm never started the study medication (Fig. 1) and were excluded from the final analysis. Sixty-five patients were finally enrolled in the study. Thirty-four received an NNRTI-based regimen and 31 received a boosted PI-based regimen. The baseline characteristics of the patients were similar between the groups (Table 1). Patients were predominantly male and were between 28 and 75 years old. For 29 out of 65 patients (44.6%), the route of HIV-1 infection had been heterosexual sex. The median CD4+ cell count was 41 cells/μl (interquartile range, 25–66.5). The median viral load was 5.54 log10/ml (interquartile range, 5.11–5.80). Almost half of the patients had previously experienced category C events.
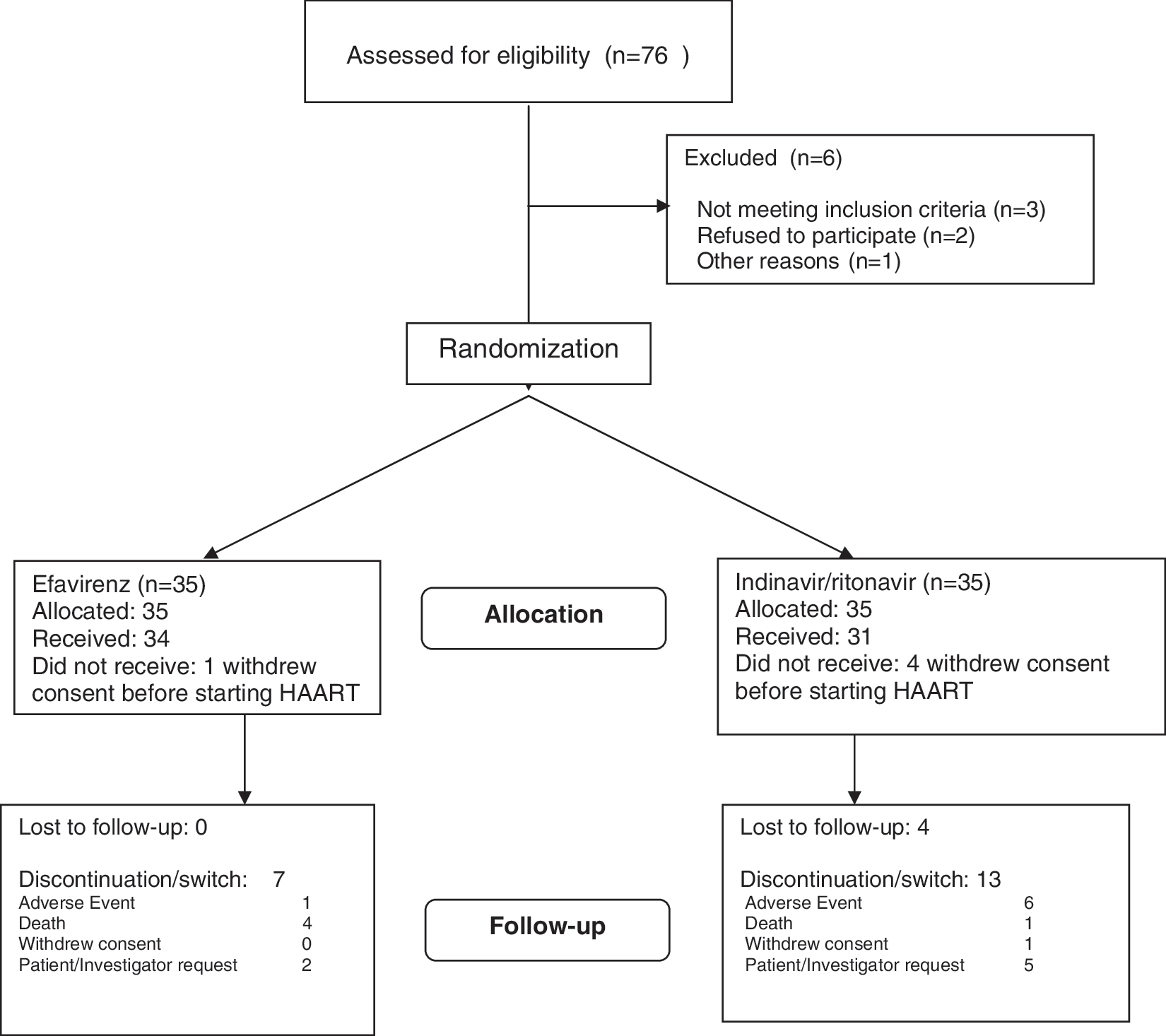
Study flow diagram.
CMV, cytomegalovirus; HBsAg, surface antigen of hepatitis B virus; HCV, hepatitis C virus; IVDU, intravenous drug users; PCP, Pneumocystis jiroveci pneumonia; PML, progressive multifocal leukoencephalopathy; TB, tuberculosis.
To convert value for glucose to millimoles per liter, multiply by 0.056. To convert value for plasma cholesterol to millimoles per liter, multiply by 0.02586. To convert value for triglycerides to millimoles per liter, multiply by 0.01129.
Immune response
By both the intention-to-treat and the on-treatment analyses, the median increase in CD4+ cell count after 12, 24, and 36 months was +199 (IQR 101–258), +226 (IQR 200–296), and +299 (IQR 170–464) cells/μl in the NNRTI arm and +136 (IQR 57–235), +168 (IQR 115–332), and +228 (IQR 119–465) cells/μl in the boosted-PI arm (p = nonsignificant for all time points by the Mann–Whitney test, Fig. 2). No differences were found between the arms in the proportion of patients reaching a CD4+ cell count above 200 cells/μl at any time point (after 12, 24, and 36 months it was 47%, 77%, and 100% in the efavirenz arm and 61%, 82%, and 84% in the boosted-PI arm). The immune reconstitution induced by an efavirenz-based regimen was at least as potent as that induced by a ritonavir-boosted protease inhibitor-based regimen at month 12 (median difference of 39 with a 95% CI of −36,115).
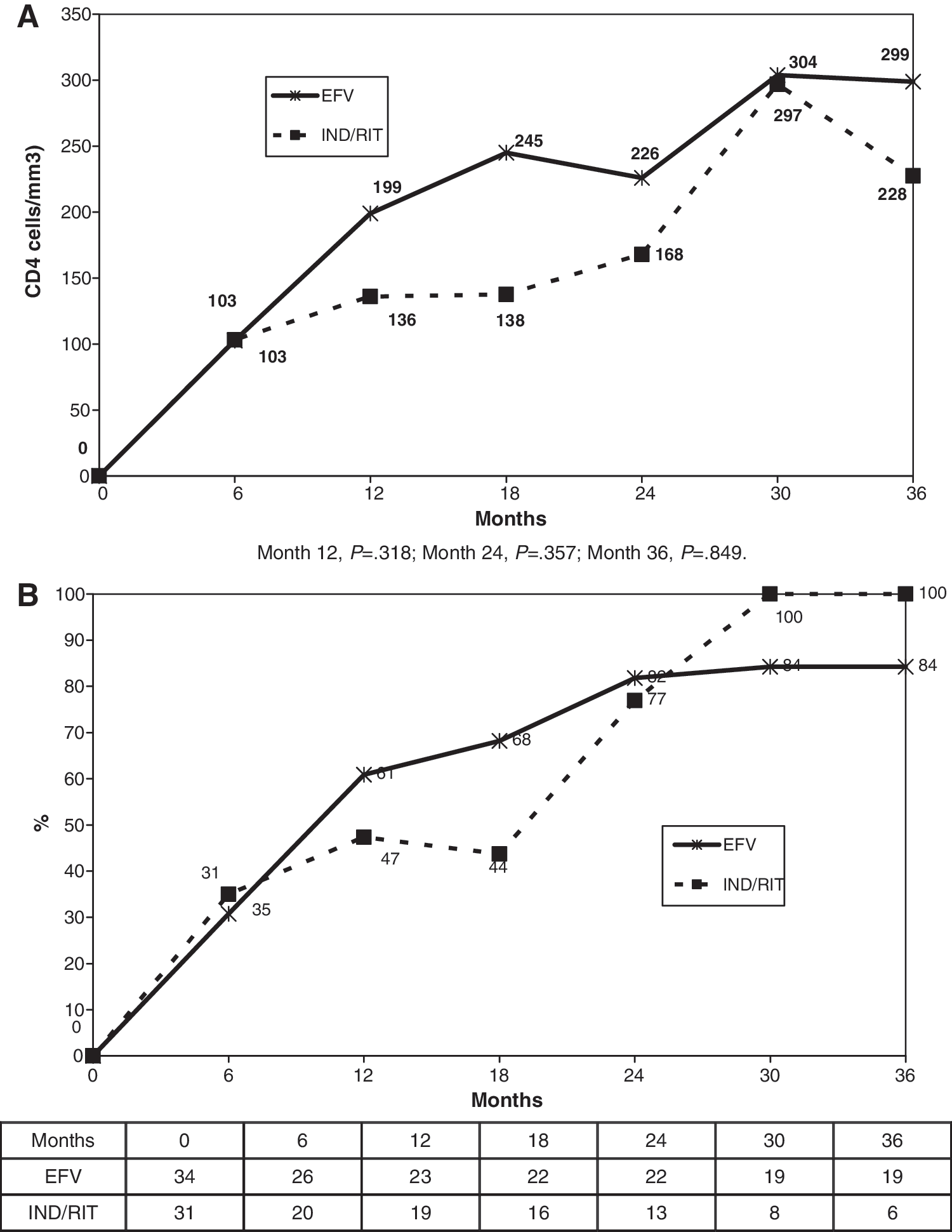
(
By the “on-treatment” analysis, there were no significant group differences at 24 months in proliferative responses to mitogens (PHA, PMW, CD3+CD28+), recall antigens, and T cell subsets (naive and memory phenotypes, CD28+ in CD4+ and CD8+ T cells) between the efavirenz and indinavir-ritonavir arms (data not shown). However, efavirenz users had significantly lower levels of immune system activation markers CD4+CD38+, CD4+CD38+DR+, CD8+CD38+, and CD8+CD38+DR+ cells (Table 2, Fig. 3).
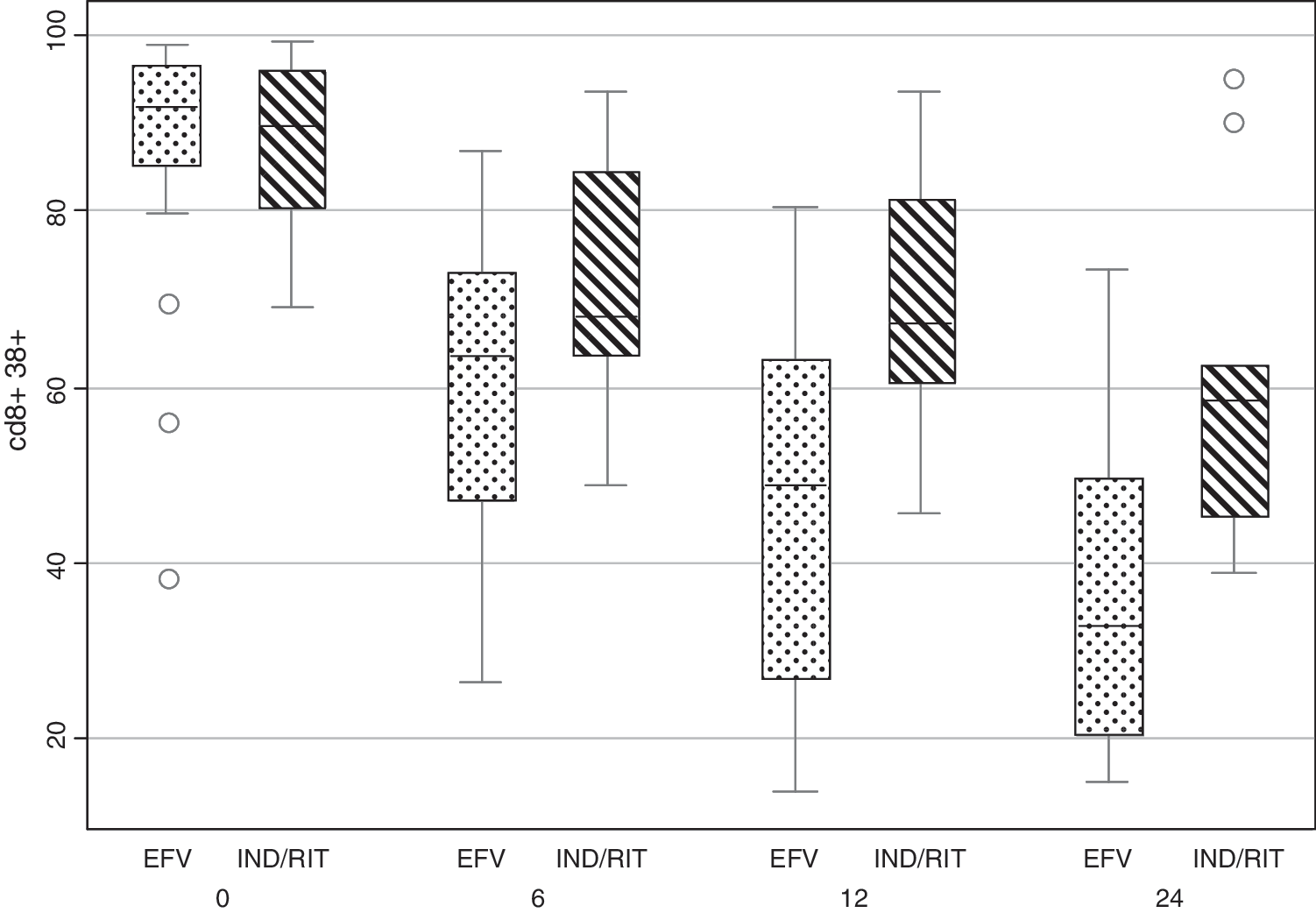
Decrease in CD38 activation markers in CD8+ T cells, by month, in the two treatment arms (on-treatment analysis). EFV, efavirenz; IND/RIT, indinavir/ritonavir. Box-whisker plot: median, interquartile range (IQR), upper/lower adjacent values (p75/p25 plus 1.5 times the IQR). Dots represent outliers (data not included between the whiskers).
EFV, efavirenz; IND/RIT, indinavir/ritonavir.
Virological and resistance outcomes
By the intention-to-treat analysis, the percentage of patients who had reached the protocol-defined endpoint of plasma viral load under 50 copies/ml at 12, 24, and 36 months was significantly higher in the NNRTI arm at 2 and 3 years, whereas no differences were found by the on-treatment analysis (Fig. 4). There were 12 virological failures (five in the NNRTI arm and seven in the boosted-PI arm) (Table 3).
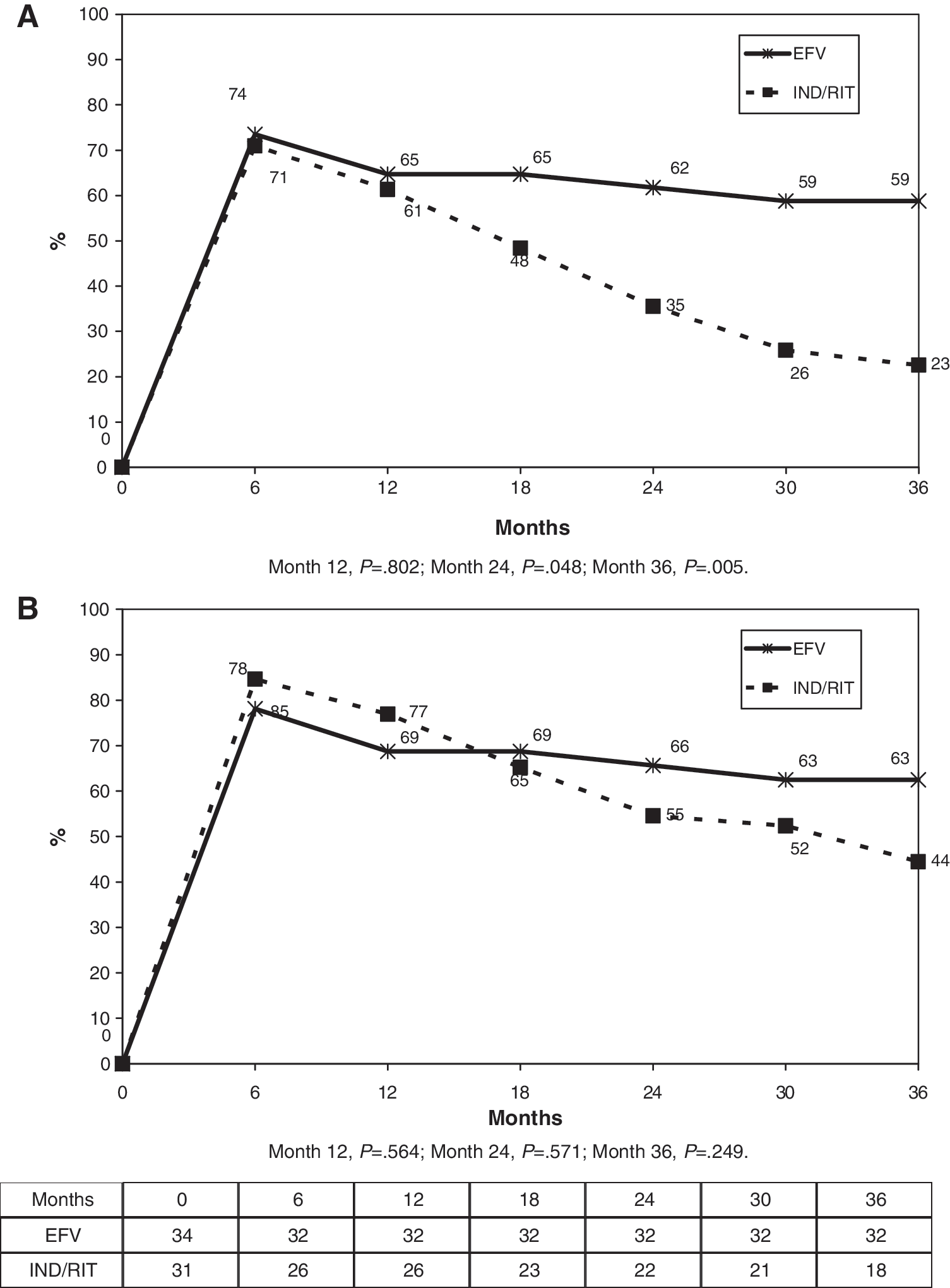
(
EFV, efavirenz; IND/RIT, indinavir/ritonavir; NRTI, nucleoside reverse transcriptase inhibitors; NNRTI, nonnucleoside reverse transcriptase inhibitors; PI, protease inhibitors; VF, virologic failure.
G190S.
L90M.
Clinical outcomes
Eleven patients (six in the efavirenz arm and five in the indinavir-ritonavir arm) developed a new CDC category C event and five patients died (four in the in the efavirenz arm and one in the boosted-PI arm) (Table 4). The Kaplan–Meier curve for new CDC category C events and death (Fig. 5) does not show any significant difference between the study arms. Fourteen patients stopped their assigned HAART (three in the efavirenz arm and 11 in the indinavir-ritonavir arm, p = 0.014) and five patients were lost to follow-up or withdrew consent (all in the boosted-PI arm). Table 4 summarizes study outcomes by the intention-to-treat and on-treatment analysis.
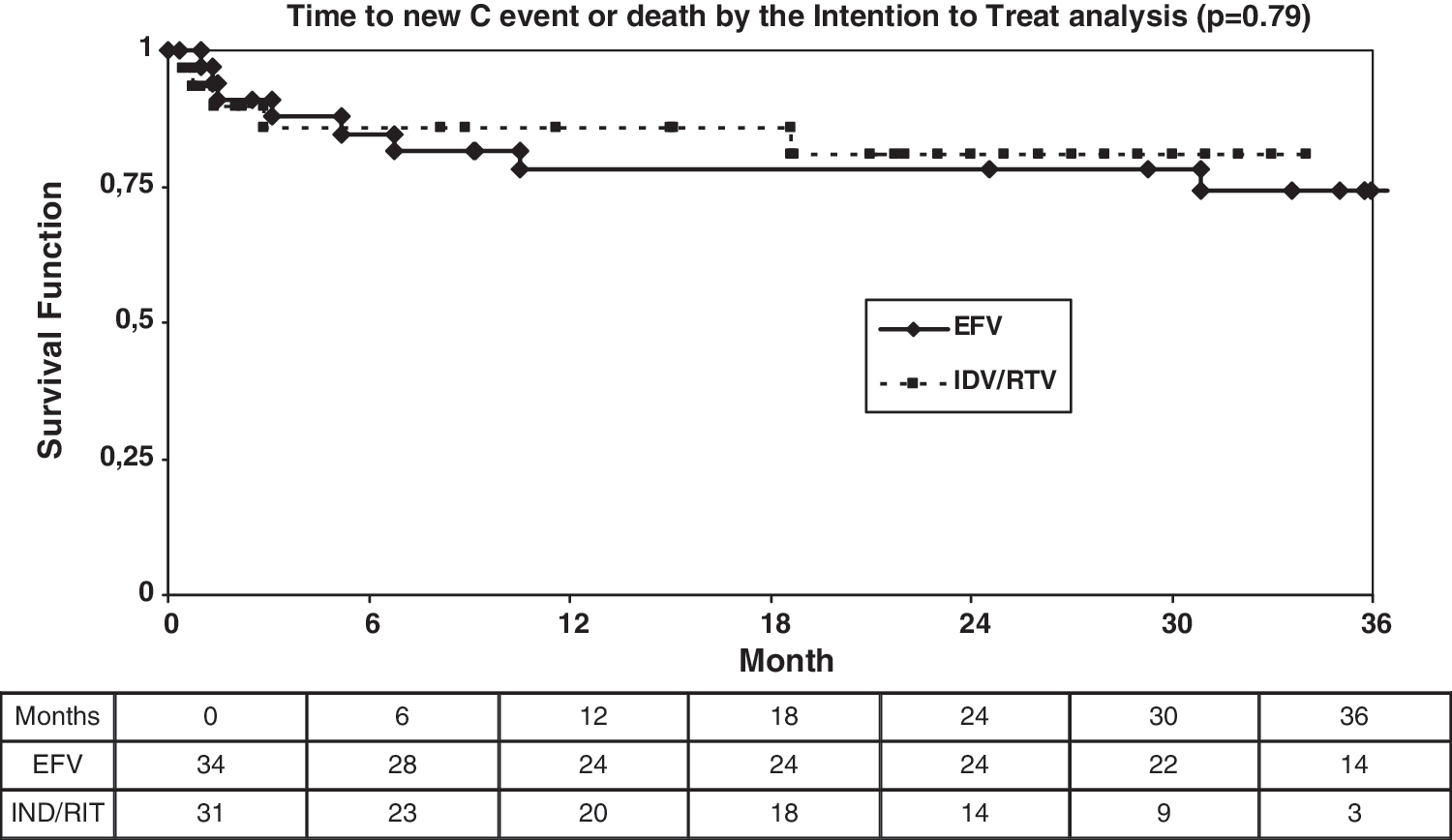
Time to new C event or death by the intention-to-treat analysis (p = 0.79 by the generalized log-rank test). EFV, efavirenz; IND/RIT, indinavir/ritonavir.
EFV, efavirenz; IND/RIT, indinavir/ritonavir; ITT, intention-to-treat analysis; OT, on-treatment analysis. Some patients had more than one event.
One Mycobacterium avium infection, one progressive multifocal leukoencephalopathy, two cervical cancer, two extrapulmonary cryptococcosis.
Two Mycobacterium avium infections, two pulmonary tuberculosis, one esophageal candidiasis.
One progressive multifocal leukoencephalopathy, one sudden death (myocardial infarction), two non-Hodgkin lymphoma.
Cirrhosis (end-stage liver disease).
Safety outcomes
The overall incidence of adverse events was significantly lower in the NNRTI group (four patients, 12%) than in the boosted-PI group (14 patients, 45%) (p < 0.01). Table 4 summarizes reported adverse events by study arm. Twenty-seven patients changed efavirenz (three switched to nevirapine and two to lopinavir-ritonavir) or indinavir-ritonavir (nine to lopinavir-ritonavir, two to atazanavir-ritonavir, eight to efavirenz, and three to nevirapine) because of adverse events.
There were no significant differences in triglyceride, total cholesterol, or glucose levels between the two groups at all time points. No significant differences from baseline were observed (p > 0.05 by the Mann–Whitney test for all time points). Zidovudine was switched in 16/65 patients (24.5%, eight in the efavirenz arms and eight in the indinavir-ritonavir arm): in 13 patients due to myelotoxicity (anemia and/or neutropenia) and in a further three patients for other reasons. In the efavirenz arm, five patients switched to stavudine, three to tenofovir, and one to didanosine; in the indinavir/ritonavir arm, four patients switched to stavudine, five to tenofovir, and one to didanosine.
Discussion
The ADVANZ study is one of the first randomized clinical trials to directly compare an NNRTI and boosted PI-based regimen in HIV-1-infected patients with very advanced disease at the time of their diagnosis. The results showed that the immune reconstitution induced by an efavirenz-based regimen at 3 years was at least as potent as that induced by a boosted PI-based regimen (indinavir-ritonavir). The antiviral efficacy was similar in both arms at 1 year by intention-to-treat analysis. However, the efavirenz-based regimen was more effective than a boosted PI-based regimen at 2 and 3 years. On-treatment analysis revealed no significant differences in efficacy between the two arms. As for virological failure, almost 50% of patients whose regimen failed in the efavirenz arm developed mutations against two classes of antiretroviral drugs (NRTIs and NNRTIs), thus confirming, as expected, a trend toward a better genetic barrier for pharmacologically enhanced PI, which allow more therapeutic options after the first failure. In both arms, almost all deaths and disease progression episodes occurred within the first year of treatment, suggesting that after this period, survivors have a good prognosis at 3 years. It is noteworthy that new CDC category C events appearing in the first 6 months after starting HAART may be due to an immune reconstitution syndrome unmasking a subjacent opportunistic infection.
From the point of view of qualitative immunological studies, most patients recovered lymphoproliferative responses to mitogens, with no significant differences between the two arms. No patients showed a response to HIV-1 recombinant glycoprotein 160 or p24, or to CMV or tetanus toxoid, according to results obtained in a small cohort study in treatment-experienced advanced patients. 16 An association between markers of immune activation and progression to clinical AIDS and death has been reported, with the intensity of CD38 staining on CD8+ T cells being the most strongly correlated parameter. Several studies have assessed the progress of immune activation with HAART and its correlation with the control of viral replication. 17 –23 The decreases in immune activation induced by the two regimens were compared. For both the PI and the NNRTI arms, the median proportion of CD4+ and CD8+ T cells expressing surface activation markers CD38 and HLA-DR decreased, but immune activation was significantly lower in the efavirenz arm from month 6 until month 24, suggesting that in very immunosuppressed patients, an efavirenz-based regimen could lead to a more reduced activation of the immune system. The intention-to-treat analysis showed that this significant reduction in immune activation in the efavirenz arm corresponded to a better suppression of HIV-1 replication at 24 months but not to a better immune reconstitution. Therefore, the clinical implications of this previously unreported finding should be explored in a more powerful study.
As for safety, efavirenz was better tolerated than indinavir/ritonavir and zidovudine was switched in almost 25% of patients due to myelotoxicity (anemia and/or neutropenia). Zidovudine is not the safest NRTI in advanced HIV-1-infected patients and other options should be considered.
A search of MEDLINE and infectious disease conference abstracts in January 2010 confirmed that very few controlled data are available on this subject. Some observational studies had previously supported the use of efavirenz-based HAART in HIV-1-infected, antiretroviral-naive individuals with very low CD4+ T cell counts, as well as good immune reconstitution and virological response. 15,24,25
An AIDS Clinical Trials Group randomized trial (A5142) 8 compared three regimens for initial antiretroviral therapy: efavirenz plus two NRTIs (efavirenz arm), lopinavir-ritonavir plus two NRTIs (lopinavir-ritonavir arm), and lopinavir-ritonavir plus efavirenz (NRTI-sparing arm). A total number of 757 patients were randomized to the three arms with a median CD4+ T cell count of 191 cells/μl and a median viral load of 4.8 log10 copies/ml. Two hundred and sixty patients (35%) had less than 100 CD4+ T cells/μl. After a median follow-up of 112 weeks, the time to virological failure was longer in the efavirenz group than in the lopinavir-ritonavir group (p = 0.006). At week 96, the proportion of patients with a plasma load of fewer than 50 copies/ml was 89% in the efavirenz arm and 77% in the lopinavir-ritonavir arm (p = 0.006). However, antiretroviral resistance mutations were more frequent in patients with virological failure in the efavirenz arm. With regard to the CD4+ T cell increase, there were no statistically significant differences at 48 weeks, although at 96 weeks, the median CD4+ T cell increase was higher in the lopinavir/ritonavir arm (287 vs. 230 cells/μl, p = 0.01). There were no specific immune reconstitution data in the group of patients with less than 100 CD4+ cells/μl.
Sierra-Madero and colleagues recently published the results from a similar trial. 26 This was a prospective, randomized, open-label trial comparing efavirenz with lopinavir/ritonavir in Mexican patients presenting with a CD4+ T cell count lower than 200 cells/μl. Baseline median CD4 cell count was 64 and 52 cells/μl in each study arm. The primary endpoint of this trial was virological. At 48 weeks, the proportion of patients achieving an HIV-1 plasma viral load lower than 50 copies/ml was 70% in the efavirenz arm and 54% in the lopinavir/ritonavir arm (p = 0.0141). In our study, no differences were found by the intention-to-treat analysis at 48 weeks (65% vs. 61%), but patients in the efavirenz arm had a higher proportion of virological suppression at 24 and 36 months. The authors showed that the mean CD4+ T cell count increase at 48 weeks was 156 cells/μl in the efavirenz arm and 168.5 cells/μl in the boosted PI arm. In our study, we found a better immunological response in the efavirenz arm (median increase of 199 vs. 136 cells/μl, respectively). However, Sierra Madero et al. 26 did not analyze the phenotypic markers of immune system activation or the response to mitogens. Although we used an earlier ritonavir-boosted PI formulation, virological, immunological, and clinical outcomes were similar when the two trials were compared.
Our study is limited by its open-label design and insufficient statistical power to detect differences in secondary endpoints between the two arms, although it did have sufficient power to detect noninferiority of efavirenz-based regimens for the immunological endpoint. Therefore, the conclusions from virological, clinical, and qualitative studies of immune reconstitution outcomes compared between the two regimens should be considered as exploratory. The superiority of the efavirenz arm by the intention-to-treat analysis is probably due to the PI formulation used. Indinavir/ritonavir was the gold standard for ritonavir-boosted PIs when the trial was started. However, this formulation is not well tolerated by patients who experience gastrointestinal and genitourinary adverse events, 27,28 which, along with the high pill burden of this formulation (adherence was not measured in our trial), caused a more frequent arm switch than in the efavirenz arm. The newer boosted PI formulations would probably be better tolerated and should be compared with efavirenz-based regimens in advanced patients. In conclusion, this study supports the use of an NNRTI-based regimen in very immunosuppressed HIV-1-infected patients. For this reason, efavirenz-based treatments can be used reliably in HIV-1-infected patients with very low CD4+ cell counts, at least in those who are expected to have a high adherence level. The availability of the new fixed-dose formulation of efavirenz, tenofovir, and emtricitabine (Atripla, one pill/day) could prove useful in this setting. Further controlled data with new antiretroviral combinations or drugs are urgently needed to increase our knowledge of the best antiretroviral treatment in advanced HIV-1-infected patients.
Appendix I: Members of the Advanz Study Group
Trial Chairs: José M. Miró, Montserrat Plana, José M. Gatell.
Trial Coordinators and Monitors: Juan A. Arnaiz, Ana Cruceta, Judit Pich.
Trial Statisticians: Elisa de Lazzari, Iñaki Perez.
Data Safety Monitoring Board: Lidia Ruiz (Hospital Germans Trias i Pujol, Badalona), Xavier Carne (Hospital Clinic, Barcelona), and Hernando Knobel (Hospital del Mar, Barcelona).
Participating Centers and Investigators (in alphabetical order): Hospital Bellvitge, Barcelona (Elena Ferrer, Daniel Podzamczer); Hospital Clínic, Barcelona (Fernando Agüero, Teresa Gallart, Felipe García, José M. Gatell, Cristina Gil, Montserrat Loncá, Christian Manzardo, Josep Mallolas, José M. Miró, Montserrat Plana, Omar Sued, Tomás Pumarola, Laura Zamora); Hospital Donostia, San Sebastian (Julio Arrizalaga, Begoña Jimeno); Hospital La Paz, Madrid (Jose R. Arribas, Alicia Lorenzo); Hospital Sant Pau, Barcelona (Pere Domingo, Montserrat Fuster); Hospital Vall d'Hebron, Barcelona (Carlos Azuaje, Esteban Ribera, Sara Villar).
Footnotes
Acknowledgments
We are indebted to the study participants. Supported in part by the “Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III, Madrid (Spain),” the Spanish Network for AIDS Research (RIS; ISCIII-RETIC RD06/006), FIS 04/0503.
J.M.M. received a Research Grant from the “Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS)” and the “Conselleria de Salut de la Generalitat de Catalunya, Barcelona (Spain).” M.P. was supported by contract FIS 03/00072 from the “Fundació Privada Clínic per la Recerca Biomèdica, Barcelona (Spain)” in collaboration with the “Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III, Madrid (Spain).”
Presented in part at the following scientific meetings: 44th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), Washington, DC (United States), October 30 to November 2, 2004, Abstract No. H-574; 10th European AIDS Conference (EACS), Dublin (Ireland), November 17–20, 2005, Abstract No. PS1/4; and 17th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID) and 25th International Congress of Chemotherapy (ICC) Munich (Germany), March 31 to April 3, 2007, Abstract No. P-1915.
ClinicalTrials.gov registration number NCT00385957.
Author Disclosure Statement
Supported in part by a research grant from Grupo Bristol-Myers Squibb, Madrid, Spain.