
Abstract
We compared HIV-1 strains in incident and prevalent infections in a cohort of men having sex with men (MSM) and female sex workers (FSW) near Mombasa, Kenya and conducted a cross-sectional study of viral isolates from a sample of HIV-1-infected MSM and FSW in Kilifi, Coast Province, Kenya. RNA extracted from plasma of 13 MSM, 9 FSW, and one heterosexual male was amplified by nested RT-PCR and the products were directly sequenced. HIV-1 strains from 21 individuals were characterized with one or more complete genome sequences, and two were sequenced in the Nef gene. The envelope quasispecies was also studied in one individual. Among MSM, eight strains were subtype A and five were recombinant. There were two epidemiologically linked pairs of sequences; one pair was subtype A and the other pair was a complex AA2CD recombinant of identical structure. Another MSM was dually infected with DG recombinant strains of related, but nonidentical, structure. MSM also harbored AC and AD recombinant strains. The FSW harbored seven subtype A strains, an AD recombinant, and an AA2D strain related to CRF16_A2D. The one heterosexual male studied had a subtype A infection. This MSM epidemic in Kenya appears to be of local origin, harboring many strains typical of the broader Kenyan epidemic. Characteristics of a close social network were identified, with extended chains of transmission, novel recombinant strains possibly generated within the network, and a relatively high proportion of recombinant and dual infections.
Introduction
HIV-1
Studies of the molecular epidemiology of HIV-1 have revealed an uneven geographic distribution of HIV strains and predominant circulating strains in the pandemic. Most infections in North America, Europe, the Pacific, and Australia are subtype B, whereas subtype C predominates in South Asia and Southern Africa. Subtype B cocirculates with CRF01_AE in Southeast Asia, with subtype C in China and subtype F in South America. 2
Africa, which has the oldest epidemic of HIV, is home to many subtypes and recombinant forms. CRF02_AG strains infect more than half of the people in the epidemic in West Africa. In East Africa, subtypes A, C, D and their recombinants, both CRFs and URFs, cocirculate in varying proportions in Uganda, Tanzania, and Kenya, although in each country a different subtype predominates: subtype D in parts of Uganda, subtype C in Tanzania, and subtype A in Kenya. An interesting feature of the Kenyan epidemic is the presence of both CRFs and URFs containing the rare subsubtype A2. 3,4
Men who have sex with men (MSM) are one of the most neglected risk populations in terms of prevention programs in many developing countries. They have become one of the populations at highest risk for HIV-1 in Latin America, African countries, Thailand, and China. 5 –11 MSM in North America, Latin America, Japan, Australia, Taiwan, and South Africa were predominately infected with subtype B strains, presumably reflecting the nature of the global HIV-1 network in developed countries. 12 –17 Ninety percent of MSM in Argentina harbored subtype B HIV-1 strains and the remaining 10% harbored subtype F strains, while 73–77% of the Argentinean heterosexual population were infected with subtype F strains. 12 Similar findings were reported in MSM in Chile 18 and in another study conducted in Equador, Peru, Bolivia, Uruguay, and Argentina. 19 Subtype B was more prevalent than subtype G among MSM in Cuba. 20 HIV-1 subtype B strains were also identified in MSM in Bogota, Colombia. 21 However, non-subtype B strains circulating locally have also been identified among the MSM population in several of these geographic regions. 5,11,22
HIV-1 subtype A1 has been the most prevalent strain in Kenya along with lower fractions of subtypes C, D, and G, and various forms of recombinants contributed substantially. 23 –26 The clear picture of the molecular epidemic in Kenya was demonstrated by full genome characterization of 41 HIV-1-positive blood donations collected from 6 hospitals across southern Kenya between 1999 and 2000. Twenty-five (61%) of the 41 strains were of pure subtype while 16 strains (39%) were intersubtype recombinants. Twenty-three strains were subtype A1 and one strain each was subtype C and D. Intersubtype recombinant A1D was predominant (15%) while A2D and A1C were equally represented (7%) in this population. A1A2D, A1CD, A1G, and CD were also identified in smaller proportions (2% each). 3 A subsequent study in Kenya by the same group evaluated HIV-1 subtypes among 60 incident infections identified in the HIV and Malaria Cohort Study conducted in Kericho from 2003 to 2006. URFs accounted for up to 55% of the characterized strains. While pure subtype A was still abundant, subtype C was rare and subtype D was not seen. One pure subtype G was identified. 27
Thus far, a detailed molecular epidemiology of MSM in Africa has not been reported outside of South Africa. 17 In this study we characterized the HIV-1 strains circulating in isolates drawn from a sample of HIV-1-infected MSM and female sex workers (FSW) participating in a cohort study in coastal Kenya in order to provide better insight into the epidemiology of HIV-1 strains circulating among potential populations for HIV-1 intervention studies in Kenya.
Materials and Methods
Study population
Since July 2005, populations in and around Mombasa, Kenya at higher risk for HIV infection, including MSM and FSW, have been targeted for recruitment into an HIV-1 vaccine preparedness prospective incidence cohort. A parallel HIV-1-seropositive cohort study was initiated in February 2006 and enrolled HIV-1-positive volunteers who were identified at screening for the incidence cohort. Study protocols were granted approval by the National Ethical Review Committee under the Kenya Medical Research Institute (KEMRI) and the University of Washington. All participants provided written, informed consent. Identification, recruitment of prospective study participants, and cohort procedures have been described elsewhere. 9 In short, peer educators identified volunteers and accompanied them to a Drop-in Centre adjacent to the research clinic. Cohort eligibility was assessed by a preenrollment counselor who discussed sexual and other risk behavior. Cohort inclusion criteria were verified by an enrollment counselor and were either a self-report of transactional sex, anal sex in the past 3 months, or same sex behavior for men. Participants committed to 3-monthly follow-up visits during a 2-year study period. At each visit, HIV-1 risk reduction counseling and testing, risk assessment by structured face-to-face interview, and medical examination with screening for sexually transmitted infections were performed. 28 Participants who seroconverted during follow up in the incidence cohort were offered enrollment into an acute HIV infection protocol. We examined samples from all seroconverters identified up to the end of year 2006 (n = 17) and a convenience sample of consecutive prevalent infections (n = 10) in MSM and FSW collected at screening during 2006.
HIV genotyping
HIV-1 RNA extracted from the plasma of infected individuals using QIAamp Viral RNA Mini Kit (QIAGEN Inc., Valencia, CA) was the source material for reverse transcription and nested DNA PCR. HIV-1 genotyping was performed in two stages. First, a screening assay, the MHAacd, was performed for each sample, as described elsewhere 29 ; this assay uses subtype-specific fluorescent probes in a Taqman real-time PCR format to determine the HIV-1 subtype in five different HIV-1 genome regions located in Gag, Pol, Vpu, and Env. In a second stage, HIV-1 characterization was performed using reverse transcriptase polymerase chain reaction (RT-PCR) and sequencing. For amplification of complete HIV-1 genomes, a two-step RT-PCR was performed using a previously described method. 30 Briefly, complementary DNA (cDNA) was synthesized from RNA, either as a complete HIV-1 genome or two half genomes overlapping by 1.5 kilobases (kb), using ThermoScript RT (Invitrogen Corp., Carlsbad, CA). JL68R (5′-CTTCTTCCTGCCATAGGAGATGCCTAAG-3′, HXB2: 5956- 5983) or UNINEF-7′ (5′-GCACTCAAGGCAAGCTTTATTGAGGCTT-3′, HXB2: 9605–9632) was used as 3′ primer to synthesize cDNA. A full genome or a half genome nested PCR was performed with near-endpoint dilution of cDNA template using a previously described method. 31 MSF12B/ UNINEF-7′ and GAG763 (5′- TGACTAGCGGAGGCTAGAAGGAGAGA-3′)/ TATANEF (5′-GCAGCTGCTTATATGCAGGATCTGAGGG-3′) were primers used in the first round and the second round to retrieve a full genome amplicon from UNINEF-7′ generated cDNA. A 5′ fragment of genome was amplified from JL68R-generated cDNA using primers MSF12B/JL68R and nested with GAG763/TAT-B′ (5′-TTCCTGGATGCTTCCA GGGCTCTA-3′). Primers POL J V2 (5′-GAAGCYATGCATGGACAAGTRGA-3′)/UNINEF-7’ followed by POLK3 (5′-TAAARYTAGCAGGAAGATGGCCAGT-3′)/TATANEF were used to amplify a 3′ fragment genome from UNINEF-7′ generated cDNA. PCR products were visualized, purified, and sequenced by Applied Biosystems 3130/3130xl Genetic Analyzers. DNA sequences were assembled using Sequencher, version 4.7. Two different half-genome sequences from an individual, with less than 0.007% bp mismatch in overlapping area of 1.5 kb, were assembled to create a full genome sequence.
For samples from which the complete genome could not be amplified, the genome region encompassing partial envelope gp 41, Nef, and partial 3′ LTR (HXB2: 8544-9524) was amplified by nested PCR from UNINEF-7′ generated cDNA with JL106 (5′- TTCAGCTACCACCGCTTGAGAGACT-3′)/UNINEF-7′ and JL106/TATANEF primers using a method previously described. 32
For one individual infected with HIV-1 subtype A, in whom two half-genome sequences showed an exceptionally high number of mismatched base pairs in the overlap region, envelope gene cloning and sequencing were performed to confirm dual infections within subtype A. The full-length envelope gene (gp160) was PCR amplified using the method previously published. 33 PCR product was cloned into a pCR-XL-TOPO vector that was subsequently transformed into MAX EFFICIENCY stbl 2 competent cells according to the instructions of the TOPO XL PCR cloning kit (Invitrogen Corp., Carlsbad, CA). Purified plasmid DNA from positive clones with inserts of expected size was sequenced.
Phylogenetic analysis
Newly derived HIV-1 sequences were aligned with reference strains of relevant subtypes and CRF. Phylogenetic analysis was performed using SEQBOOT, DNAPARS, DNADIST, NEIGHBOR, and CONSENSE modules from the PHYLIP package, version 3.4. Bootscanning and informative site analysis were performed to identify and verify pure subtype viral strains and to identify and map intersubtype recombinants. 34,35 Visual inspections of multiple sequence alignments were also performed to precisely map breakpoints in recombinant strains, and subregion trees were constructed along with bootstrap values to further confirm subtype assignments of the different segments of recombinant genome structures.
Results
Study participants
The Kilifi-Coast prospective incidence cohort study included 211 MSM and 148 FSW until the end of 2006. Seventy-six percent of MSM participants reported transactional sex in the previous 3 months. Over 96% of MSM and 98% of FSW were from Kenya. The HIV-1 incidence in MSM has been very high and more than double the incidence in FSW. 9 Table 1 provides an overview of HIV-1 genotyping results for 23 individuals. Thirteen subjects were MSM; among these, eight reported sex with men exclusively, while six reported sex with both men and women. Five MSM were HIV-1 seropositive at study entry (prevalent infections) and eight were HIV seronegative at baseline but became HIV-1 infected during follow-up (incident infections). Nine female sex workers (FSW) were studied, four with incident HIV-1 infection and five with prevalent infections. One study subject was a male reporting only heterosexual exposure and an incident HIV-1 infection.
MSM, men who have sex with men; FSW, female sex workers; HM, heterosexual male.
Individuals reporting sex with both males and females.
FL, full genome sequences.
HIV-1 genotyping
Out of the 23 samples, 22 were first characterized using a fluorescent genotyping assay and 14 showed reactivity only with subtype A probes. Eight samples showed evidence of intersubtype recombination, including strains AC, AD, and ACD. Because of the presence of many intersubtype recombinants, complete genome sequencing was selected as the optimal approach for HIV-1 genotyping.
Complete genome sequences were obtained for 21/23 of the study subjects, including all 13 MSM, 7 FSW, and the heterosexual male (HM). For two of the FSW, full genome sequencing was not successful, and sequencing was limited to a portion of the Nef gene. Multiple, complete genomes were sequenced from two of the MSM, and multiple sequences of the complete envelope gene, in addition to a full-length sequence, were obtained from one of the FSW samples. Table 1 records the GenBank accession numbers for these sequences, and summarizes the overall subtyping results based on the sequence data. Two individuals were found to harbor more than one strain or subtype, indicated by “dual” in Table 1.
Nonrecombinant HIV-1
Figure 1 presents the results of phylogenetic analysis of the HIV-1 sequences. Fourteen strains were nonrecombinant, and all clustered with the subtype A reference sequences (Fig. 1A, large bracket) with a bootstrap value of 100%. Careful examination of these strains by bootscanning confirmed that they were subtype A throughout the genome (data not shown). Figure 1A shows that the subtype A strains from MSM (brown) and from the FSW and the HM (green) were intermixed in the tree, and roughly equidistant. The one exception was strains 06KECst_005 and 06KECst_022, both from MSM; these were much more closely related, and clustered with bootstrap values of 100% (small bracket, Fig. 1A). This relationship is consistent with direct transmission between these two individuals. In summary, 17 of 23 isolates studied (74%, including 8 of 13 MSM, 7 of 9 FSW, and one HM) harbored pure subtype A strains.

HIV-1 subtypes and recombinant forms in Mombasa, Kenya. Analysis of strains without evidence of recombination are presented in
The sequence of strain 06KECst_013, from a prevalent HIV-1 infection in an FSW, was initially found to be quite heterogeneous. Accordingly, the full-length (FL) sequencing was performed at a higher template dilution to obtain an unambiguous complete genome sequence and the diversity of the viral quasispecies in Env was simultaneously explored by molecular cloning. Both the FL sequence (Fig. 1A) and the cloned Env (gp160) sequences (Fig. 1B) were subtype A. Among the envelope clones, two distinct clusters, each supported by 100% bootstrap values and consisting of three and five sequences, respectively, were observed. These data are consistent with a dual subtype A infection in this subject.
Figure 1C shows that the partial Nef sequences from FSW 06KECst_018 and FSW 06KECst_024 are also subtype A.
Intersubtype recombinant HIV-1
Eight strains from seven individuals were intersubtype recombinants. All were characterized by full-genome sequencing. Figure 1D shows their subtype composition and structure, the breakpoints between segments of different subtypes, and the bootstrap values supporting the subtype assignments of the different segments. The subtypes of these segments, and their positions in the HIV-1 genome, numbered according to reference strain HXB2, are shown in Table 2. Four of the MSM harbored intersubtype recombinants, as did two of the FSW. Strain 06KECst_004, from an MSM, was an AC recombinant. It was subtype A throughout the genome except for the Vpu gene, which was subtype C. Strains 06KECst_008 and 06KECst_011, from two MSM who were sexual partners, are complex AA2CD recombinants of identical structure by available mapping techniques. The A2 subsubtype constitutes the majority of the genome, with segments of subtype D in Gag, Pol, and Env. Subtype A segments are found in Pol, in two regions of Env, and in Nef. There is also a small subtype C segment in Pol. The interstrain nucleotide distance between these two recombinants is less than 1% (data not shown). Strain 06KECst_015 is an AD recombinant, mostly subtype A with two segments of subtype D in Pol.
Numbering according to reference strain HXB2.
Bootstrap value (%) joining this segment with the indicated subtype.
Not determined; the right half-genome of this strain was not recovered among five clones analyzed.
The fourth MSM with a recombinant strain, 06KECst_014, was dually infected, and harbored the first complete DG recombinants described to date. From this individual, the HIV-1 genome was amplified and sequenced as left- and right-half genomes, overlapping by 1.5 kb. In the left half of the genome, two molecular forms were observed, represented by two and three sequences, respectively. The six right-half genomes were identical in structure and, in the overlap region, matched only one of the molecular forms, the one represented by two sequences. The Form 1 structure shown in Fig. 1D represents the assembly of these matching halves, and the Form 2 structure depicts the remaining three left-half genomes of a different structure. Both of these molecular forms were DG recombinants. Form 1 is mostly subtype G, with a segment of subtype D in the Pol gene. Form 2 differs in two respects from form 1: the subtype D segment in Pol is shortened at its 3′ end, and an additional subtype D segment is found in the Vif gene. Right-half genomes corresponding to Form 2 were not recovered among the sequences analyzed. Thus, this study participant harbors at least two DG recombinant strains of related structure.
Two intersubtype recombinant strains were found in FSW (Fig. 1D). Strain 06KECst_027 was a complex AA2D recombinant and strain 06KECst_010 was a subtype A strain with the p17 region of Gag from subtype D.
CRF16_A2D-related strains
Subsubtype A2, thought to have originated in West Africa, is present within the HIV-1 strains from three of the seven individuals with recombinant HIV-1 strains in this study. The pair of complex A2-containing strains linked by transmission (06KECst_008 and 06KECst_011, respectively) do not share any obvious breakpoints in common with the A2-containing FSW strain (06KECst_027). Since all of these strains contain segments of subtype D, for which strains from East and West Africa are phylogenetically distinct, we also performed a separate analysis of some of their larger subtype D regions. The MSM strains contained East African subtype D, but the subtype D segments within the AA2D strain from the FSW were from West Africa. Figure 2 presents a complete analysis of strain 06KECst_027 with respect to other A2-containing strains previously reported from Kenya.
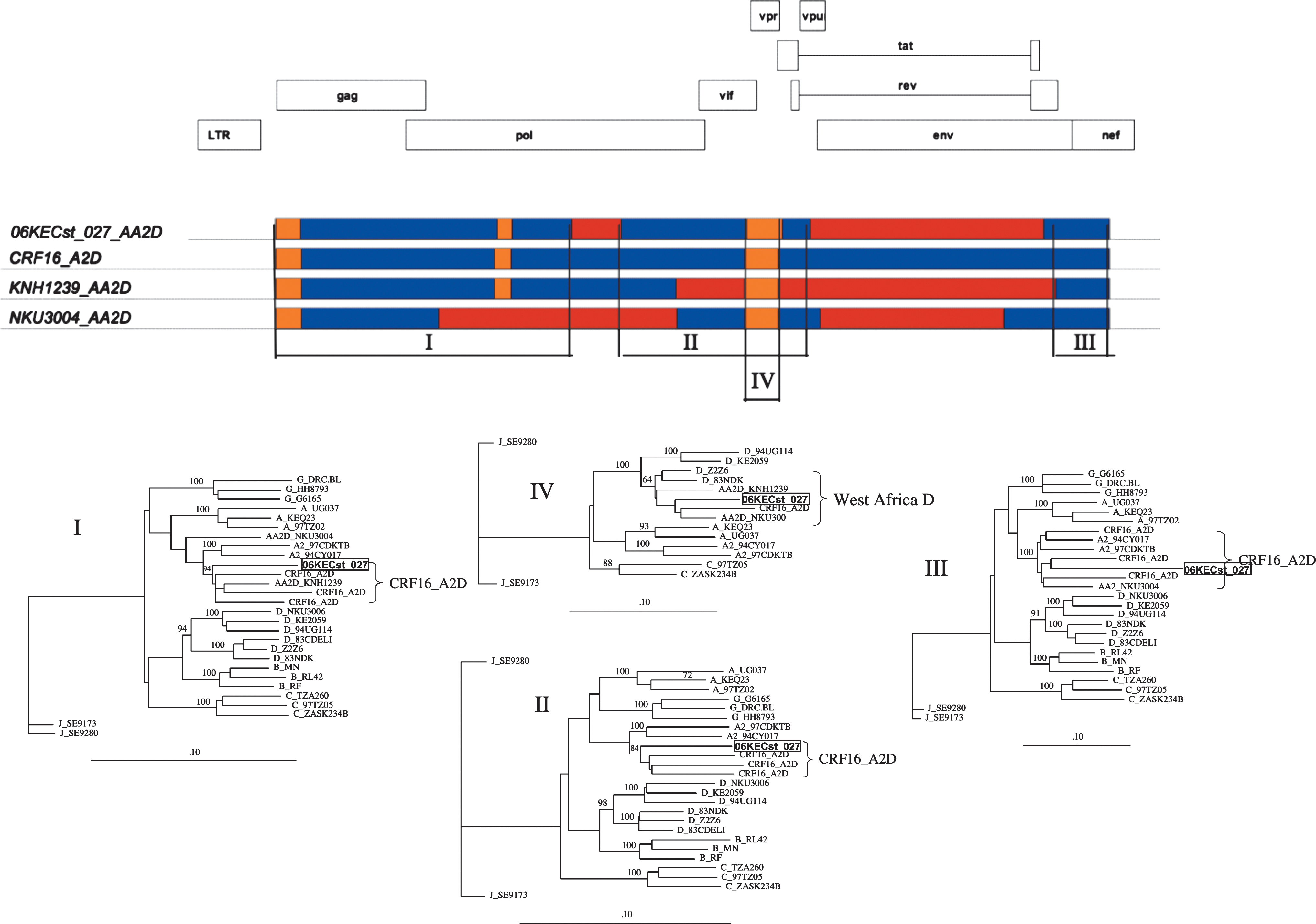
Recombinants related to CRF16_A2D. Top: The structure of strain 06KECst_027, an AA2D recombinant from an FSW in Mombasa, Kenya, is compared to CRF16_A2D, and to two other AA2D strains previously reported from Kenya. Subtype A, red; A2, dark blue; D, orange. Genome regions I–IV were selected for further analysis. Bottom: Phylogenetic trees establishing the relationships among strains, constructed using neighbor joining with bootstrapping. Scale bar represents a 10% difference. Bootstrap values at important nodes are indicated. Regions I, II, III, and IV correspond to genome positions 796–3774, 4275–6178, 8640–9180, and 5532–5891 in HXB2, respectively. The clustering of 06KECst_024 with CRF16_A2D (regions I–III) and with West African subtype D strains (region IV) is indicated by brackets.
Strain 06KECst_027 has the structure of CRF16_A2D in some genome regions, but is subtype A in other parts of the genome. Therefore, CRF16_A2D, and two previously reported AA2D recombinants, also related to CRF16_A2D, were included in the analysis. We analyzed four informative subregions (I–IV, Fig. 2). The subregion trees for region I, from HXB2 position 796 to 3774, and from region II (HXB2 4275–6178) both show 06KECst_027 clustering specifically with CRF16_A2D, with which it shares an identical structure in these regions (bootstrap values 95% and 84%, respectively). In region III, this strain, along with CRF16_A2D and the other AA2D recombinants, cluster with the A2 reference sequences. In region IV, all four of these strains have a subtype D Vpr, which clusters with West African, rather than East African, subtype D reference strains (bootstrap value 64%).
Overall, this analysis shows that most of strain 06KECst_027 is derived from CRF16_A2D, with a subtype A region in pol and most of the envelope from subtype A. This new strain is not obviously related to KNH1239_AA2D or to NKU3004_AA2D, previously described in Kenya. However, all three AA2D recombinants related to CRF16_A2D have most, if not all, of their envelope from subtype A.
Eight new intersubtype recombinant forms have been identified in this study. Table 2 shows the genome location of the breakpoints separating segments from different subtypes, and the bootstrap values supporting the subtype classification of the different segments. All of the bootstrap values exceeded 70%, the commonly accepted threshold for significance, except for two regions of about 200 bp in the related AA2CD strains 06KECst_008 and 06KECst_011, assigned to subtype C and A2, respectively, and a 142-bp segment assigned to subtype D in strain 06KECst_027; the informative sites in these segments support the indicated subtype assignments, but the segments are too short to attain significance in phylogenetic trees.
The sequences from this study are under GenBank accession numbers FJ623475 to FJ623505.
Discussion
This MSM epidemic in Coastal Kenya appears to be of local origin, harboring many strains typical of the broader Kenya epidemic. Full genome characterization revealed that HIV-1 strains circulating in Kenyan MSM contained genetic materials of subtype A1, A2, C, D, and G as previously reported in other Kenyan populations. 3,24 –27,36 Pure subtype A was predominant in both MSM and in FSW. Recombinant strains composed of various combinations of the locally circulating subtypes, namely AC, AA2CD, AD, and DG, accounted for 5/13 circulating strains in MSM. A similarly complex molecular picture was also seen in FSW, with AA2D and AD recombinants (3/9). No subtype B was found in this population, suggesting a local origin of HIV-1 in Kenyan MSM. This contrasts with the described MSM epidemics in South Africa 17 or South America 12,37 where subtype B is predominant in MSM but local strains (subtype C in South Africa and BF recombinants in South America) were mostly identified in heterosexuals. These findings confirm behavior studies on 81 MSM from the same cohort, the majority of whom were sex workers. These MSM sex workers engaged in anal and vaginal sex with women and both receptive and insertive anal sex with men providing transmission bridges to the general heterosexual population. 38 The subtype data confirm these observed behavioral links between MSM and the general population.
In this relatively small study, prevalent infections in MSM were subtype A, AD, and DG recombinants while prevalent FSW infections were subtype A only; this may indicate an ongoing higher degree of complexity of HIV-1 in MSM populations compared to FSW. The diversity in the MSM group was underscored by the existence of a recombinant between subtypes D and G that has been identified for the first time in this Kenyan MSM cohort. This suggests that HIV-1 risk(s) in MSM may be different from that in FSW, and that a higher proportion of recombinant strains among incident infections in Kenyan MSM was one of the consequences.
Incident infections always reflect the most current HIV-1 molecular picture in the epidemic and although limited by the small sample size, we nevertheless observed that in both MSM and FSW incident infections harbored more recombinant strains than the prevalent infections. AC and AA2CD were identified in MSM and AD and AA2D in FSW. Both populations harbored relatively high proportions of recombinant strains among incident infections: 38% of MSM and 50% of FSW, respectively. The complexity of the molecular epidemic here was similar to the results from a recent HIV-1 incident infection study conducted in 2006 revealing 50% recombinant strains circulating in a low-risk population in Kericho, Kenya. 27 Taken together with the report that more than half of these MSM have sex with women as well as men, the evidence of direct connection between MSM and the local community is strong.
Among MSM there were two subtype A strains identified from two infected individuals, 06KECst022 and 06KECst005, clustered closely in the tree. This revealed a close social network with extended chains of transmission as seen in other high-risk populations such as intravenous drug users cohorts in Thailand. 39 Kenyan MSM also shared this characteristic since two similar complex recombinant strains among subtypes A1, A2, C, and D were identified from two different MSM individuals.
Another unique characteristic of multiple subtype epidemics is dual infections, thought to represent the proximal source of new recombinant strains. HIV-1 subtype A dual infection was identified among FSW as evidenced by the two distinct A clusters of multiple envelope sequences in subject 06KECst_013. In addition, two different forms of DG recombinants identified in an MSM individual also suggest that this Kenyan MSM population harbors dual infections and is a potential source of transmission with recombinant strains.
In Kenya, CRF16_A2D, CRF21_A2D, and other A2 containing recombinants were previously identified. 3,4 Strain 06KECst_027, an AA2D recombinant identified in an FSW individual, shares some breakpoints with CRF16_A2D, KNH1239_AA2D, and NKU3304_AA2D in some genome regions. A complete analysis of these recombinants revealed that the newly identified 06KECst_027 is clearly related to, and possibly derived from, CRF16_A2D. This suggests an ongoing process of recombination, since the introduction of A2-containing strains in a high-risk population in Kenya, similar to that seen among MSM in China. 11
In conclusion, this MSM epidemic in Coastal Province, Kenya appears to be of local origin, harboring many strains typical of the broader Kenya epidemic and infections in local FSW. It consists of a close social network with extended chains of transmission, contains novel recombinant strains possibly generated within the network, and has a high proportion of recombinant and dual infections. The Coastal Kenyan MSM population presents a key target for public health and behavioral HIV prevention interventions.
Footnotes
Acknowledgments
We are grateful to the subjects for their participation in the study. We thank the clinic staff of the KEMRI-clinic in Mtwapa, the laboratory staff at the KEMRI-Wellcome Trust Research laboratories in Kilifi, Meera Bose for technical assistance, and Eric Sanders-Buell for his advice on HIV-1 cloning and sequencing. The study was supported by the International AIDS Vaccine Initiative (IAVI). The opinions or assertions contained herein are the private views of the authors, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense, the Kenya Medical Research Institute, or IAVI. This article was published with permission from the Director of KEMRI.
Author Disclosure Statement
No competing financial interests exist.