
Abstract
We report the near full-length genome characterization of an HIV-1 subtype F virus (D88_845) collected in St. Petersburg, Russia, from a 25-year-old Russian woman perinatally infected in 1982. In a Bayesian phylogenetic analysis, the genome sequence branched basally to the subsubtype F1 clade. In partial sequences, D88_845 clustered with 13 other subtype F sequences from Russia, corresponding to gag (n = 2), pol (n = 3), and env (n = 8) segments. At least 11 of these sequences are from samples collected in St. Petersburg from heterosexually infected Russian individuals. In each of these segments, the Russian viruses formed a monophyletic cluster that branched as a sister clade of the F1 subsubtype. One sequence from Belgium branched with D88_845 with a posterior probability of 0.99. This is the first report on the identification and near full-length genome characterization of the subtype F variant circulating in St. Petersburg, which is closely related to, but distinct from, the F1 subsubtype.
Russia is the European country with the greatest number of HIV-1 infections. 1 Currently the HIV-1 epidemic in Russia is dominated by the former Soviet Union subtype A (AFSU) variant, transmitted mainly among injecting drug users (IDUs), with a more recent secondary spread via heterosexual contact. 2,3 This variant, which began to spread epidemically among IDUs in early 1995 in the southern Ukrainian city of Odessa, derives from an older strain that was transmitted heterosexually at a low prevalence in southern Ukraine and originated in central Africa. 4 Before the beginning of the IDU epidemic, HIV-1 infections in Russia were identified mainly among homosexual men, infected with subtype B, sub-Saharan African men and their local heterosexual contacts, harboring diverse African clades, and children in southern Russia infected in a nosocomial outbreak with a subtype G strain.
Previously, we reported the HIV-1 subtype distribution in 341 samples collected from 1996 to 2004 in different areas of Russia, based on protease-reverse transcriptase (PR-RT) sequences, of which 271 (79.5%) were of subtype A, 48 (14.1%) of subtype B, 9 (2.6%) of CRF03_AB, 5 (1.5%) of subtype C, 3 (0.9%) of subtype G, 2 (0.6%) of subtype F, 2 (0.6%) of CRF01_AE, 2 (0.6%) were unique AB recombinants, and one was a dual B + C infection. 3,4 In a more recent study, on 102 samples collected in 2006 in St. Petersburg, also based on PR-RT sequences, we found that 93 were of AFSU, 8 of subtype B, and 1 of subtype F. 5
Here, we report the analysis of the near full-length genome sequence of a fourth subtype F sample collected in St. Petersburg in 2008, examining its relationship to other subtype F viruses from Russia and elsewhere.
Sample D88_845 was collected in 2008 from a 25-year-old woman, perinatally infected in 1982 from her Russian mother, who presumably acquired her HIV-1 infection via sexual contact from a former sexual partner from the Democratic Republic of Congo (DRC). These are probably the earliest documented cases of HIV-1 transmission in St. Petersburg. 6 In addition, we have obtained a newly derived near full genome sequence of a virus of subtype F in PR-RT, collected from a Spanish woman, residing in Galicia (Northwestern Spain) infected via heterosexual contact, for whom there are no other epidemiological data available.
The near full-length genome of D88_845 was amplified from plasma RNA by RT-PCR and sequenced as previously described. 7,8 X845_4 was similarly amplified from RNA extracted from the culture supernatant of a primary isolate. Sequences were aligned using MAFFT v.6.240. 9 Phylogenetic analyses were performed with the Bayesian Markov Chain Monte Carlo (BMCMC) method as implemented in MrBayes 3.1. 10 The nucleotide substitution model used for the analysis was the general time reversible with gamma-distributed rate heterogeneity across sites and a proportion of invariant sites (GTR + Γ + I). Two simultaneous independent runs were performed, with eight chains, sampling every 500 generations. Support for the nodes was derived from a majority rule consensus of 1000 trees from the posterior distribution sampled after both runs had reached convergence, as determined by an average standard deviation of split frequencies <0.01. The possible existence of recombination was analyzed by the bootscanning method. 11 For this analysis we used a window of 600 nucleotides moving in 100 nucleotide increments, and trees were constructed via maximum likelihood with PhyML 12 applying the GTR + Γ + I model of substitution.
In a Bayesian phylogenetic tree, which included all near full-length genomes of subtype F available in public databases 13 (Fig. 1), D88_845 joined the subsubtype F1 clade in a basal position with a 1.0 posterior probability (PP). Within the subsubtype F1 clade, viruses formed two strongly supported subclades. One included viruses from Brazil and Argentina, together with one virus from Finland (FIN9363), probably acquired in East Africa, and one from Belgium (VI850) with epidemiological links to the DRC. The second cluster included two viruses of Romanian origin (X1670 and P1145, from Romanians residing in Spain), one of Angolan origin (X1093_2, from an Angolese woman residing in Spain), one (MP411) from a French individual who had traveled to Chad and former Yugoslavia, and X845_4. The last sequence branched closely with the Angolan virus with a PP of 1.0.
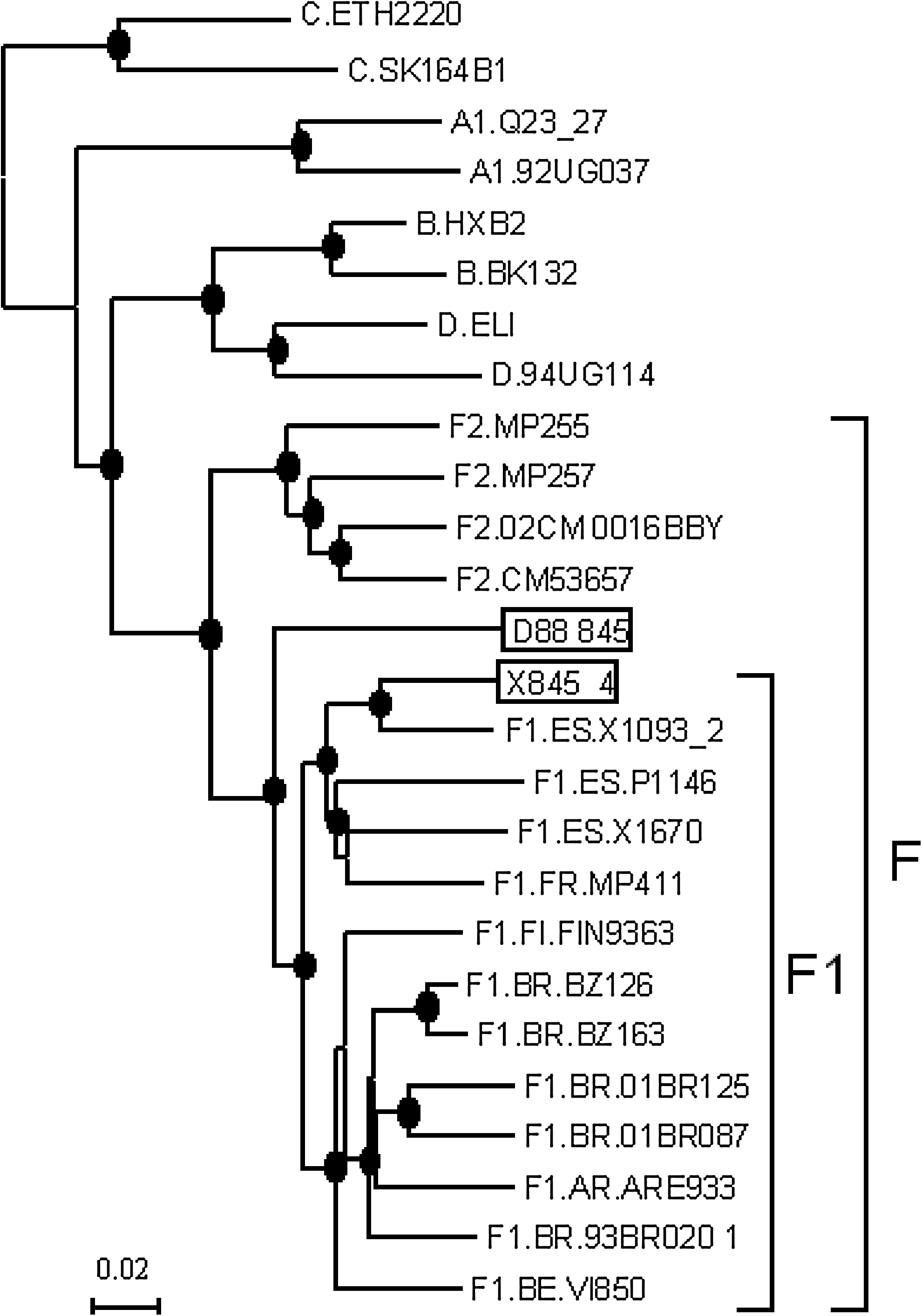
Phylogenetic tree of the near full-length genome sequence of D88_845 and X845_4. Trees were constructed with the BMCMC method using MrBayes v3.1. Nodes supported by a PP = 1.0 are marked with a filled circle.
In bootscan analyses, D88_845 and X845_4 grouped uniformly with the subsubtype F1 references all along their genomes, although D88_845, in a short segment at the 5′ end of integrase, in which F1 and F2 failed to form separate clusters, joined the F1/F2 clade with only a 57% bootstrap value (results not shown).
We constructed a tree of PR-RT sequences including D88_845 and three other subtype F sequences from St. Petersburg, previously obtained by us from samples collected in 2004. 4,5 All four Russian F sequences formed a monophyletic clade, supported by a 1.0 PP, which branched as a sister clade of the F1 references (Fig. 2a). All three previously obtained sequences corresponded to Russian heterosexually infected individuals without known epidemiological links to each other or with D88_845. RUSP_B_059 was from a woman diagnosed with HIV-1 infection in 1994; RUSP_813 corresponds to a woman infected between December 1991 and March 1993, who reported previous sexual contacts with five men, all of them Russian; and RUSP816 was from a man who first tested HIV-1 seropositive in 1993.
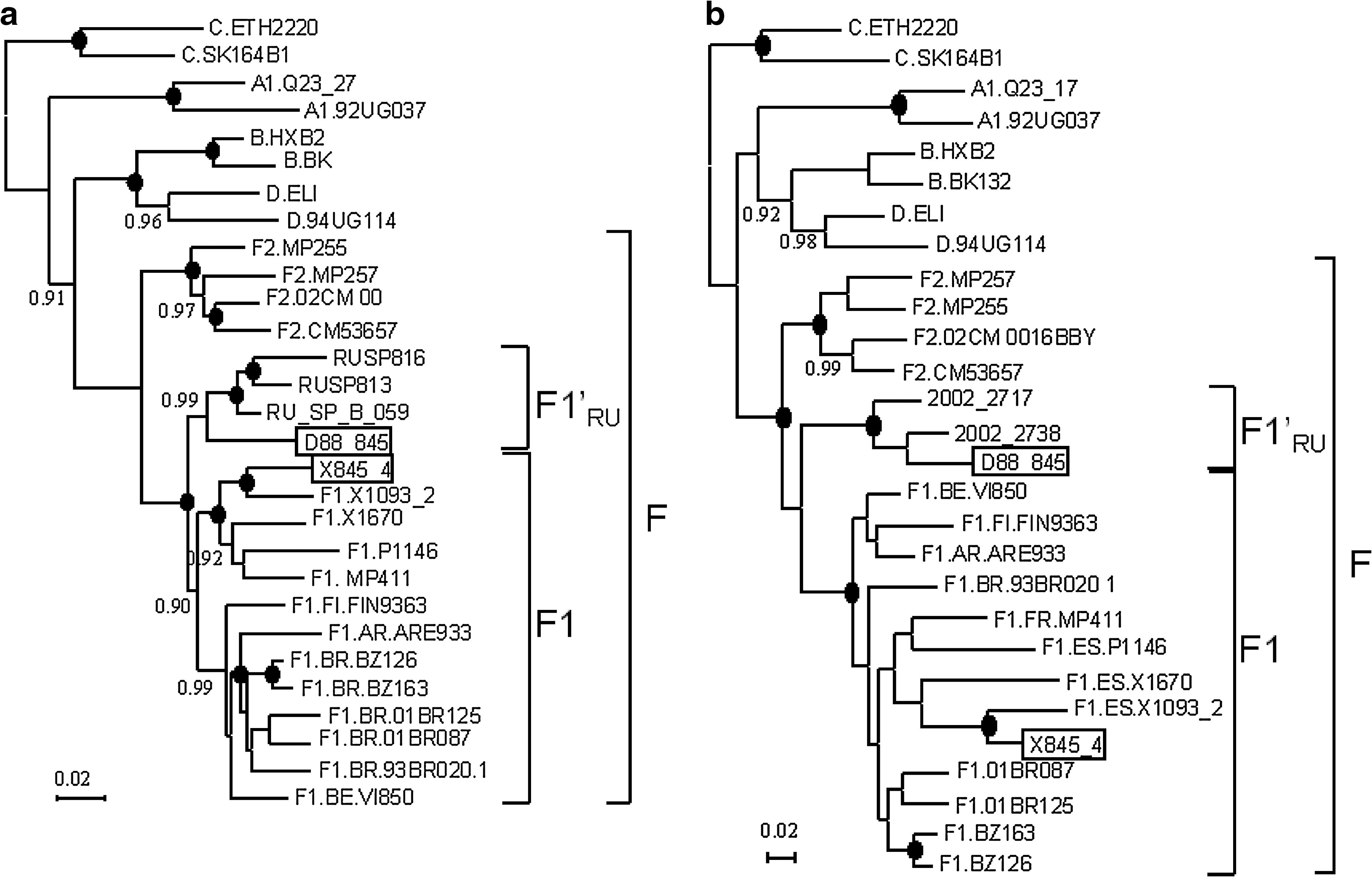
Phylogenetic trees of partial sequences, showing the relationships of Russian subtype F viruses. Trees were constructed with the BMCMC method using MrBayes v3.1. Nodes supported by a PP = 1.0 are marked with a filled circle. PP = 0.90–0.99 are shown on the left of the corresponding nodes. F1′RU denotes the clade formed by Russian F viruses. (
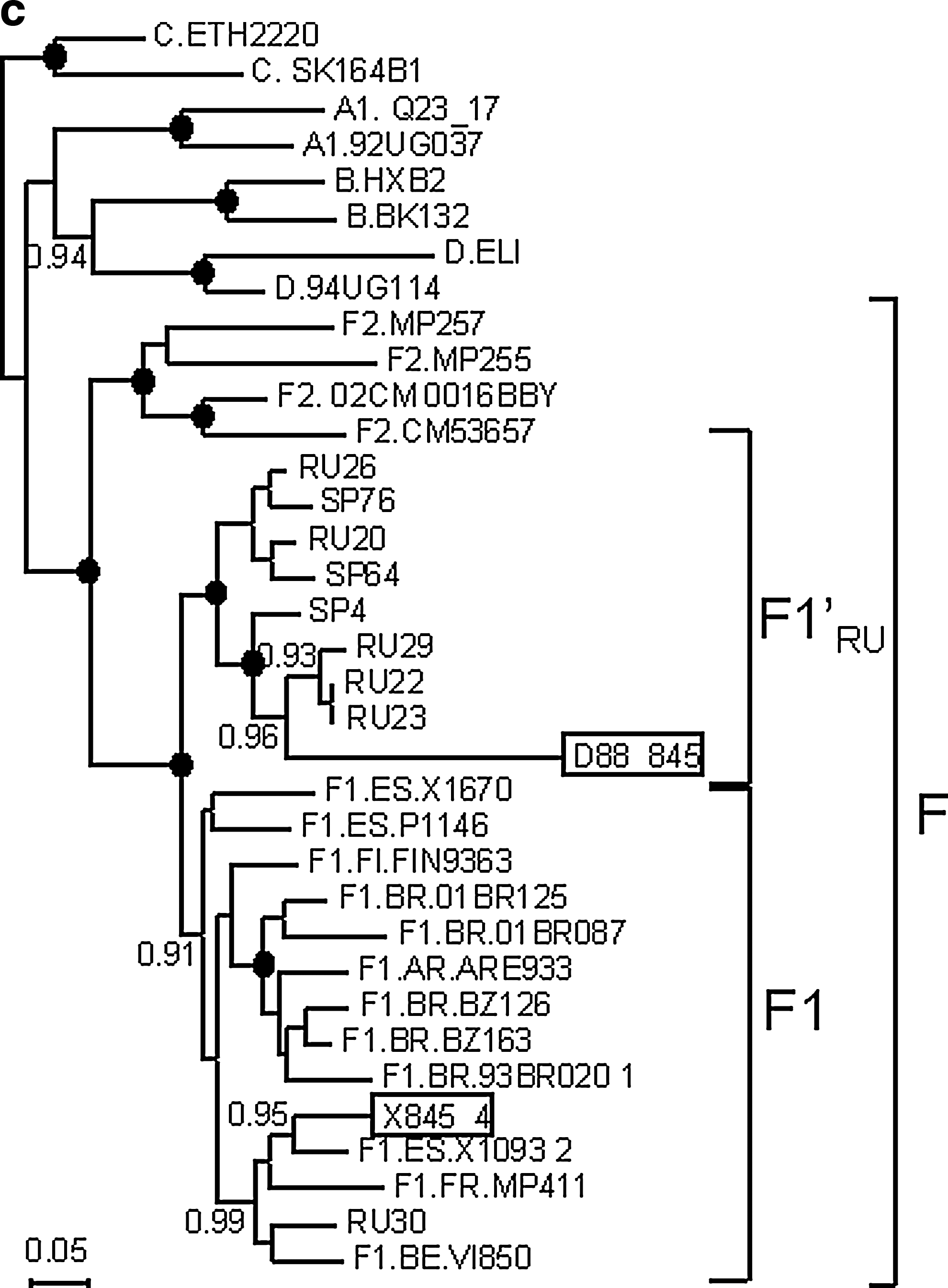
Phylogenetic trees were also constructed with 11 other subtype F sequences from Russia obtained by other authors and deposited in databases, nine corresponding to the env C2-V3 region 14,15 and two to gag. 16 All but one of the Russian subtype F sequences formed strongly supported monophyletic clusters, which included D88_845, in both segments, branching in each one as a sister clade of the F1 clade (Fig. 2b and c). The only subtype F virus collected in Russia branching outside of the Russian F cluster (R30) was from a Congolese individual. 14 Of the 10 gag or env sequences branching within the Russian F cluster, it is known that at least eight are from samples collected in St. Petersburg from heterosexually infected Russian individuals 14,15 and the other two are from the northwestern Federal District of Russia, which comprises St. Petersburg. 16
To determine the origin of the F strain circulating in Russia we performed BLAST searches of all F sequences retrieved from the Los Alamos HIV Sequence Database against all full-length F1 references, including D88_845. Those with highest similarity scores to the Russian virus were subjected to BMCMC phylogenetic analysis. Only one virus from Belgium branched with D88_845, with a PP value of 0.99 (Fig. 3).
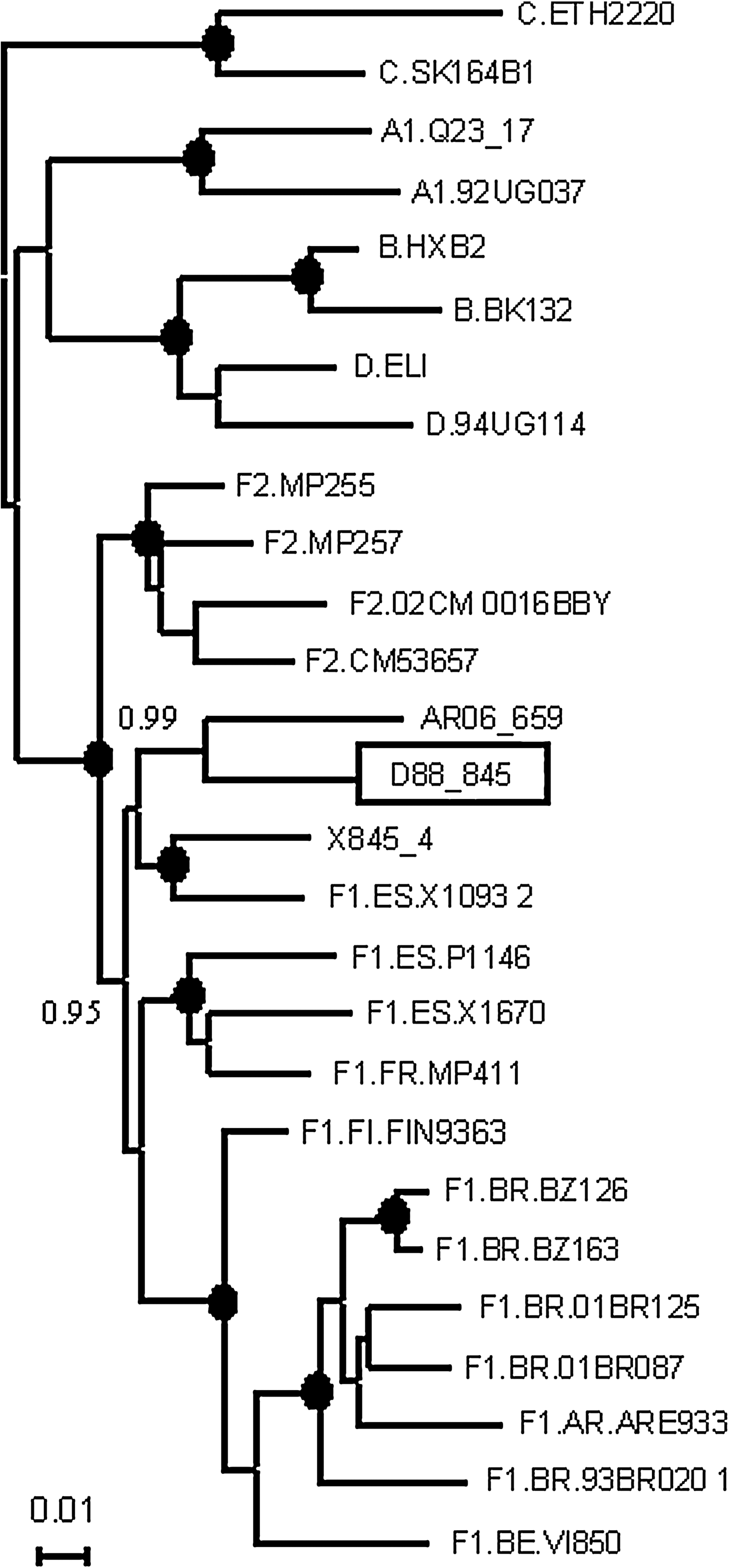
Phylogenetic tree showing the relationship of D88_845 with a virus collected in Belgium (AR06_659). The analyzed segment corresponds to HXB2 nt positions 4230–5096. The tree was constructed with the BMCMC method using MrBayes v3.1. Nodes supported by a PP = 1.0 are marked with a filled circle. PP = 0.90–0.99 are shown on the left of the corresponding nodes.
The results of the study, therefore, support the existence of a distinct subtype F variant of monophyletic origin circulating in Russia, closely related to subsubtype F1 but branching as a sister clade relative to the F1 radiation, rather than inside it, indicating a common ancestry with the F1 subsubtype. We will designate this variant F1′RU, to reflect its close relationship to the F1 subsubtype, although, considering its topological position, it is not strictly an F1 variant.
In addition, we report the analysis of a newly derived near full-length genome sequence of a subsubtype F1 virus (X845_4) from a Spanish woman, which is closely related to an F1 virus from Angola, suggesting a probable epidemiological link with this country.
F1′RU is transmitted at a low prevalence via heterosexual contact in St. Petersburg, where at least 11 of 13 F1′RU viruses were collected. It is probable that the origin of this variant is the former sexual partner of the mother of D88_845, a man from the DRC. Contact-tracing investigations revealed that after being diagnosed with HIV-1 infection, the mother of D88_845 transmitted HIV-1 to at least one man, who subsequently infected at least three other women. Thus, the F variant might have spread via heterosexual contact from the former sexual partner of the mother of D88_845 to at least five persons, who could have further propagated the virus in St. Petersburg.
In a previous study, based on C2-V3 sequences, it was reported that five Russian subtype F viruses, also analyzed in this study, formed two independent clusters, suggesting their origin from two separate introductions in Russia. 14 The discrepancy with our results, which support a single introduction, may derive from different methods used for phylogenetic inference, a neighbor-joining method with Kimura two-parameter distances in the previous study and a BMCMC method with a GTR + Γ + I model of evolution used by us.
Among non-Russian database sequences, only one from Belgium branched with D88_845 with strong node support. Considering that in Belgium there is a large immigrant population from the DRC, a former Belgian colony, and that D88_845 probably derives from a man from the DRC, we can postulate that this country is the most likely origin of the ancestor of the Russian F subtype variant. It is interesting to note that the DRC also appears to be the country of origin of the AFSU variant, 4 which dominates the HIV-1 epidemic in most former Soviet Union countries
Although the prevalence of F1′RU is currently very low in Russia (1% in a recent survey in St. Petersburg 5 ), it may be important to be aware of its existence and to monitor its prevalence in view of reports on the rapid expansion of minor variants when introduced in preexisting transmission networks 4,17 and of reported differences of subtype F viruses in susceptibility to some antiretroviral drugs 18,19 and in drug resistance mutation profiles compared to other subtypes. 20
Footnotes
Acknowledgments
We thank Aurora de Miguel, Ana Parejo, and Miguel Ovejero from the Genomic Unit at the Centro Nacional de Microbiología, Instituto de Salud Carlos III, Majadahonda, Madrid, Spain, for technical assistance in sequencing. This work was funded through a technical service agreement with WHO-UNAIDS OD/TS-07-00446 and through the European network of excellence EUROPRISE (LSH/CT/2005/037611).
Sequences have been deposited in GenBank, with accession numbers GQ290462 and FJ670516.
Disclosure Statement
No competing financial interests exist.