
Abstract
Cells of the immune system express a number of receptors that bind carbohydrate ligands. We questioned whether peptide mimetics of these ligands will activate phagocytic cells and thereby enhance an antiviral response. Short peptide sequences were identified by computational modeling of docking to glycan-specific lectins, selected as receptor analogs, and incorporated into quadravalent structures by peptide synthesis. A peptide with the sequence HPSLK bound to several lectins specific for monosaccharides and to lectins specific for Neu5Ac-Gal-containing complex glycans, whereas a longer sequence, NPSHPLSG, bound only lectins specific for the more complex glycans. In cultures of peripheral blood mononuclear cells (PBMCs) these peptides stimulated phagocytosis of opsonized microspheres. The peptides inhibited replication of HIV-1 in PBMC cultures by 20–80% at concentrations between 1 nM and 1 μM but inhibited replication 100% in the presence of diluted HIV-positive antiserum that alone inhibited replication by 30%. HPSLK caused about 50% loss of viability of cells at 1 mM, a concentration 106-fold higher than an effective inhibitory concentration, but no toxicity was observed with NPSHPLSG. These results demonstrated that peptidomimetics of glycan ligands of cellular receptors are effective in activating phagocytosis, which may be a factor in providing complete inhibition of HIV-1 replication in vitro.
Introduction
HIV-1,
Phagocytic cells are regulated by an extensive array of cell surface receptors. Some are C-type lectins, which bind sugars in a calcium-dependent manner. 14,15 A C-type N-acetylgalactosamine (GalNAc)-binding receptor, CD301, is expressed on the surface of macrophages and immature dendritic cells and is involved in endocytosis. 16,17 C-type lectins that also undergo endocytosis include DC-SIGN and the mannose (Man) receptor, which bind Man; Langerin, which binds galactose (Gal); and Dectin-1, a β-glucan receptor. 15 Other receptors are the I-type lectins that belong to the immunoglobulin superfamily. The best characterized members of I-type lectins are siglecs (sialic acid-binding Ig-like lectins), which bind sialic acid (5-acetylneuraminic acid, Neu5Ac)-Gal-containing glycans and modulate signaling events in the immune system. 18,19 Several siglecs, particularly CD22 (siglec-2), 20 CD33 (siglec-3), 21 siglec-5, 22 and sialoadhesin (CD169), 18,19,23 undergo endocytosis on binding of sialylated ligands. Whereas most siglecs contain an inhibitory immunoreceptor tyrosine-based inhibitory motif (ITIM) sequence in their cytoplasmic domain, 18,19 siglec-14 and siglec-15 lack this domain and function in conjunction with an activating adaptor protein, DAP10 or DAP12, which contain an immunoregulatory tyrosine-based activating motif (ITAM) domain. 24,25
Glycans have several drawbacks as therapeutic agents, in particular, their difficult synthesis and stability. Consequently, peptide mimetics of sugars are gaining interest as substitutes. 26 We questioned whether short peptides would mimic sugar components of natural ligands of receptors that function to stimulate phagocytosis. Peptide mimetics of sugars have potential advantages over glycans and glycoproteins because of chemical synthesis and ease of purification. Moreover, peptide-based structures can be constructed that bind with higher affinities to lectins than glycan ligands. 27 A number of peptides that mimic sugars have been identified, some of which closely resemble specific sugars 28 –30 and others that act as more general mimetics. 31 Some peptides can functionally mimic a sugar, such as those with the consensus core sequence YPY that inhibit the mitogenic activity of the Man-specific lectin concanavalin A (Con A), yet bind at a site different from the saccharide-binding site. 32,33 Peptide mimetics have been studied as vaccines to elicit antibodies against sugar antigens, including those on the surface of HIV, 27,31 and complex oligosaccharides. 34,35
We designed several short peptide sequences by computational modeling that were predicted to have significant affinity to lectins that have the sugar specificity of several receptors. These sequences were incorporated into multivalent structures to achieve clustering of ligands and receptors, features that are required for high-affinity interactions, 19 and to accommodate the possibility that cross-linking of receptors is required for activation of phagocytosis. 36 Well-characterized plant lectins were used as receptor analogs to analyze the mimetic properties of the peptides. In binding assays, the peptide with sequence HPSLK had characteristics of a general sugar mimetic and bound to several lectins with higher affinity than their natural ligands. Highest affinities were found with lectins that bind monosaccharides such as Neu5Ac, GalNAc, or fucose. Longer peptides, in particular NPSHPLSG, did not bind these lectins but bound strongly to lectins specific for di- or trisaccharides that terminate with Neu5Ac-Gal. Because these oligosaccharides are components of complex glycan ligands that bind specific receptors, particularly siglecs, we determined the ability of the peptides to activate phagocytosis of adherent cells in cultures of human peripheral blood mononuclear cells (PBMCs). Nanomolar concentrations of the peptides dramatically stimulated uptake of opsonized microspheres. The peptides had modest ability to inhibit HIV-1 replication in PBMCs, but complete inhibition was achieved in the presence of antibodies against the virus.
Methods and Materials
Peptide design and synthesis
Unique peptide sequences were designed by computer modeling of docking to sugar-binding sites of lectins, downloaded from the Protein Data Bank, with ArgusLab 4.0.1 software (Mark A. Thompson, Planaria Software LLC, Seattle, WA,
Peptides were also synthesized with the sequences NPSHPSLG and NPSHPLSG. Peptides (200–300 mg) were dissolved in water, neutralized with Na2CO3, applied to a column (1 × 5 cm) of CM-Sephadex C-50, and washed extensively with water to remove side-products of synthesis. Peptides were eluted with 0.1 N HCl, neutralized, and then purified on a preparative Jupiter Proteo C12 column (21.2 mm × 250 mm) (Phenomenex, Torrance, CA) using a gradient of acetonitrile in water containing 0.1% trifluoroacetic acid (TFA). The eluted peptides were dried under vacuum, dissolved in water, and then passed through a DEAE-Sephadex A-25 column (1 × 30 cm) to remove TFA and endotoxin, diluted into 100 mM NaCl, and filter sterilized. Concentration was determined by the bicinchoninic acid assay (Pierce, Rockland, IL) using the dansylated peptide (extinction coefficient, ɛmM = 5.7 cm−1 at 336 nm) as standard. Correct synthesis was confirmed by mass spectroscopy and amino acid sequence analysis.
Lectin binding
For lectins available as peroxidase conjugates, 50 μl of 2 μM biotin-tagged quadravalent peptide in phosphate-buffered saline (PBS), pH 7.2, was added to each streptavidin-coated well of a microtiter plate (binding capacity, 125 pmol per well, Pierce) and incubated 1 h at room temperature. The wells were blocked with 1% gelatin in 50 mM Tris–HCl, pH 7.5, and washed two times with 50 mM Tris–HCl (pH 7.5) containing 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, and 1 mM MnCl2 (buffer A). Then 50 μl of 1 μg/ml horseradish peroxidase-conjugated lectins (Sigma-Aldrich) in buffer A were added. After 1 h incubation, wells were washed four times with buffer A and then 50 μl peroxidase substrate (1-Step Ultra TMB-ELISA, Pierce) were added. After 2–5 min the reaction was stopped with 50 μl 2 M H2SO4 and absorbance was read immediately at 450 nm. The amount of lectin bound was calculated from the specific activity of the peroxidase-conjugated lectins (OD450/min/ng protein).
The protocol was modified to assay binding of peptides to unconjugated lectins from Sambucus nigra (SNA1) and Maackia amurensis (MAA). Lectin-coated microwell strips (AlerCHEK, Portland, ME) were hydrated in buffer A, blocked with 1% gelatin in buffer A, and then 100 pmol of biotinylated peptide were added to each well. After 1 h incubation, the wells were washed three times with buffer A and then 50 μl of 0.3 μg/ml peroxidase-conjugated streptavidin (Sigma-Aldrich) were added. Wells were washed four times with buffer A and peroxidase activity was assayed as above.
Phagocytosis assay
Human PBMCs were purchased from Cellular Technology Ltd. (Shaker Heights, OH) and cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS). Dragon green-labeled, streptavidin-coated microspheres (0.97 μm diameter, Bangs Laboratories, Inc., Fishers, IN) in 0.1% bovine serum albumin and 0.05% Tween 20 were incubated with biotinylated recombinant HIV-1 envelope gp41 (ProSpec-Tany Technogene LTD, Rehovot, Israel), washed with PBS, opsonized with polyclonal rabbit anti-gp41 (ProSpec), and washed again with PBS. Alternatively, microspheres were incubated with a 1:1 dilution of polyclonal rabbit antistreptavidin serum (Sigma-Aldrich) and washed three times with PBS. PBMCs were incubated 20 h with 50 nM peptide in microtiter plates and then microspheres were added at approximately a 10:1 ratio of microspheres to cells. After an hour of incubation, formalin was added to a concentration of 2%, the samples were allowed to stand at 4°C overnight, and were then washed three times with PBS to remove free microspheres and examined with an inverted microscope. Unfixed cells were analyzed for phagocytosis of the fluorescent microspheres, opsonized with the antistreptavidin serum, by flow cytometry with a 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany). Cytochalasin D was used as an inhibitor of phagocytosis. As the reference, SYTO®62, a red fluorescent nucleic acid stain, was used and only doubly labeled cells were counted as phagocytic.
Cytokine assays
Human PBMCs (Cellular Technology Ltd) were plated at 4 × 105 cells per well in RPMI 1640 medium containing 10% FBS. After 2 days at 37°C in 5% CO2, peptide or lipopolysaccharide (LPS), as a positive control, at final concentrations of 100 nM and 10 ng/ml, respectively, was added and the incubation continued for 4 h. Media were then withdrawn and cytokines assayed in duplicate with the 40-cytokine human inflammation antibody array by RayBiotech, Inc. (Norcross, GA).
HIV replication assays
HIV-1 replication was assayed in cultures of T (MT2) cells as described previously. 39 HIV-1 strain 23135, isolated by Carl Hanson at the California Department of Public Health, and peptide at several concentrations between 0.1 nM and 1 μM were added to the culture and cells were immobilized under agarose. After 6 days at 37°C, the cells were stained with propidium iodide and plaques were counted.
PBMCs were prepared through Ficoll gradients from buffy coats purchased from the American Red Cross blood bank under IRB approval held by the blood bank. Aliquots of 5–10 × 107 cells were stored frozen in 90% FBS + 10% DMSO in liquid N2. Cells were thawed, sedimented, suspended in RPMI-1640 medium containing 25% FBS and 5% interleukin (IL)-2 (ZeptoMetrix Corp. Buffalo, NY), and activated with 5 μg/ml phytohemagglutinin (PHA) for 24 h at 37°C in 5% CO2. Cells were washed free of PHA, suspended in RPMI-1640 medium containing 10% FBS and IL-2, and then added to a 96-well microtiter plate (50 μl, 2.5 × 105 cells/well). Peptide (100 μl) was added followed by 50 μl of medium or diluted HIV-positive serum, pooled from North American AIDS patients (final dilution 1:360). R5 HIV-1 strain SF162 (clade B) or 97ZA009 (clade C) was then added (100 μl, 100 TCID50) and replication assayed according to a standard protocol. 40,41 SF-162 was obtained through the NIH AIDS Research and Reference Reagent Program from Jay Levy 42 and HIV-1 97ZA009 through the NIH AIDS Research and Reference Reagent Program from Dr. Robert Bollinger and the UNAIDS Network for HIV Isolation and Characterization. Cells were incubated 3 days at 37°C, then washed three times to remove free virus, peptide, and antiserum and suspended to 250 μl of medium. After an additional 24-h incubation, cells were lysed with Triton X-100 and protein p24 in each sample was assayed by ELISA to quantify the amount of virus.
Determination of cytotoxicity
Viability of cells was determined after staining with acridine orange and ethidium bromide. 43,44 In this assay, viable cells show green fluorescence whereas dead cells show red fluorescence. Percent viability was determined by the number of viable cells in peptide-treated cultures divided by viable cells in cultures with vehicle alone.
Results
Identification and synthesis of glycan mimetics
We previously reported 45 that the sequence VQATQSNQHTPR, identified during a screen of a phage display library with the lectin from Helix pomatia (HP), behaves as a mimetic of Gal/GalNAc. Preliminary experiments indicated that this peptide stimulated phagocytosis of bacterial cells (L.L. Eggink and J.K. Hoober, unpublished data). To further explore sugar mimetics as activators of phagocytes, additional, shorter peptides were designed by computational modeling of docking to several lectins whose structures and sugar-binding sites are well characterized. We initially chose lectins specific for Gal, GalNAc, and Neu5Ac. HPSLK was identified as an amino acid sequence predicted to bind these lectins with a highly favorable binding energy.
The ability of peptides to bind to the lectins was determined with a solid-phase assay. The HPSLK peptide was synthesized as monovalent, bivalent, or quadravalent constructs, with the active sequence extended from a GGGS linker sequence attached to a mono-, di-, or trilysine core, respectively. The C-terminus was extended with ɛ-biotinyl-lysine to anchor the peptide to streptavidin in wells of a microtiter plate. After extensive washing, lectin retained in the wells was detected by peroxidase activity conjugated to the lectin. As shown in Fig. 1, strong binding to the qudravalent peptide was found with lectins from Dolichos biflorus (DB), specific for GalNAc
46
; Triticum vulgaris (wheat germ agglutinin, WGA), specific for Neu5Ac, N-acetylglucosamine (GlcNAc),
47,48
and clusters of GalNAc
49,50
; and Ulex europaeus (UEA1), specific for
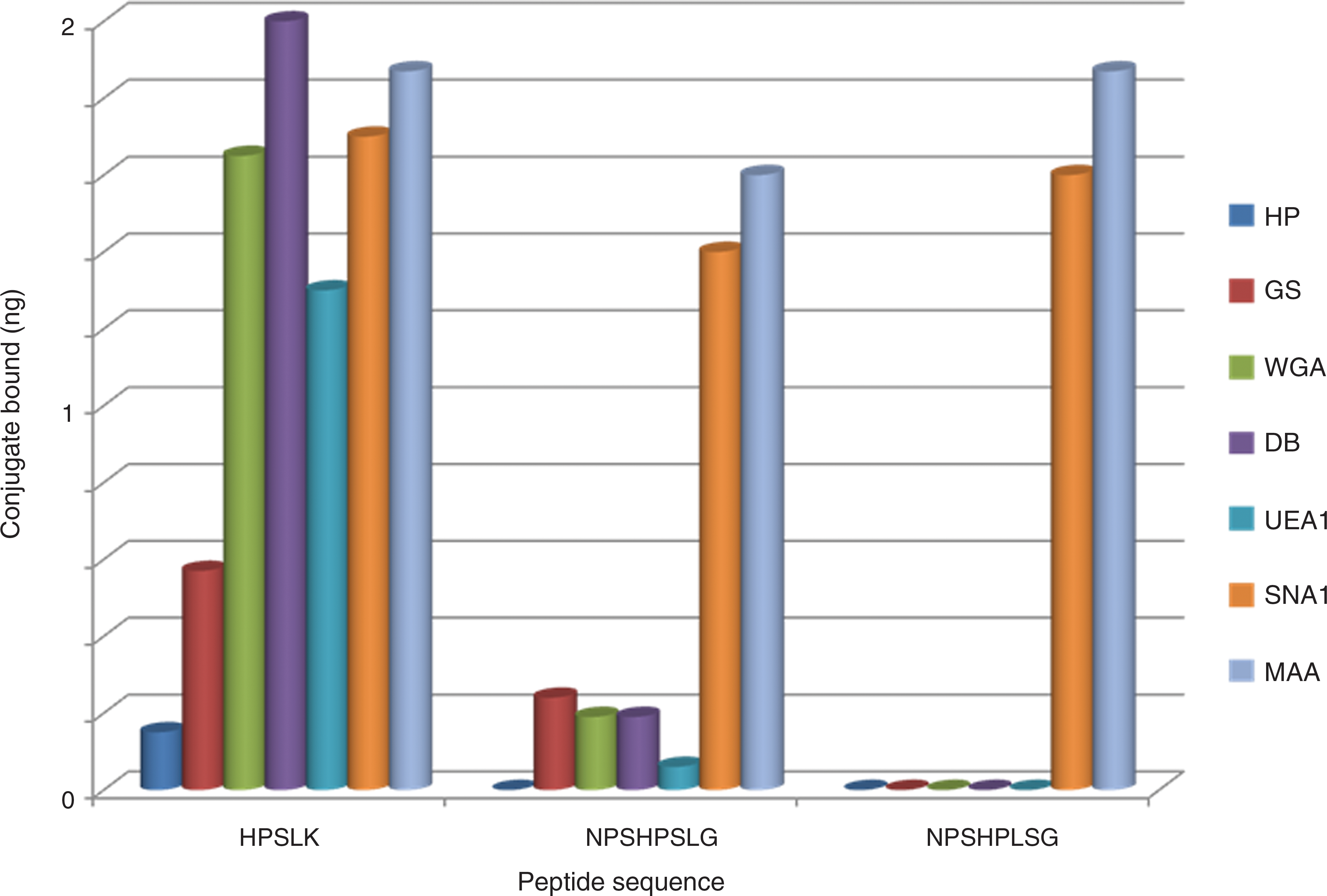
Binding of quadravalent peptides, containing the sequences shown, to lectins. Dark blue, HP; red, GS; green, WGA; purple, DB; blue, UEA1; orange, SNA1; light blue, MAA.
We were particularly interested in whether multivalent structures show enhanced binding to the lectins over a monovalent peptide, as predicted by entropic factors 54,55 and ligand density. 31,56 In an assay with equal numbers of sequences, a bivalent structure bound to lectins about 50% as strongly and the monovalent peptide about 10% as strongly as the quadravalent peptide (Fig. 2). Quantitative competition binding assays were performed to estimate affinity of the HPSLK peptide to lectins. Neu5Ac (neutralized) was used as competing monosaccharide with WGA, which binds to the lectin with a K d value of 1 mM. 57 A 50% inhibition of binding of the bivalent peptide was obtained at 90 mM Neu5Ac and of the quadravalent peptide at about 300 mM Neu5Ac. A K d of 22 nM was calculated from these data for binding of the bivalent molecule and of 7 nM for the quadravalent peptide to WGA. Only 20% inhibition of binding of HPSLK to DB was obtained with 250 mM GalNAc. To test for inhibition by more complex glycans, porcine gastric mucin, which contains glycans with GalNAc, Gal, GlcNAc, and fucose, with a minor amount of Neu5A, as terminal sugars 58,59 was added. Mucin (50 μg/ml) inhibited binding of the HPSLK peptide to HP, DB, WGA, and UEA1 by greater than 80% (data not shown). These competition experiments support the conclusion that the peptides bound with high affinity to the glycan-binding site of the lectins.
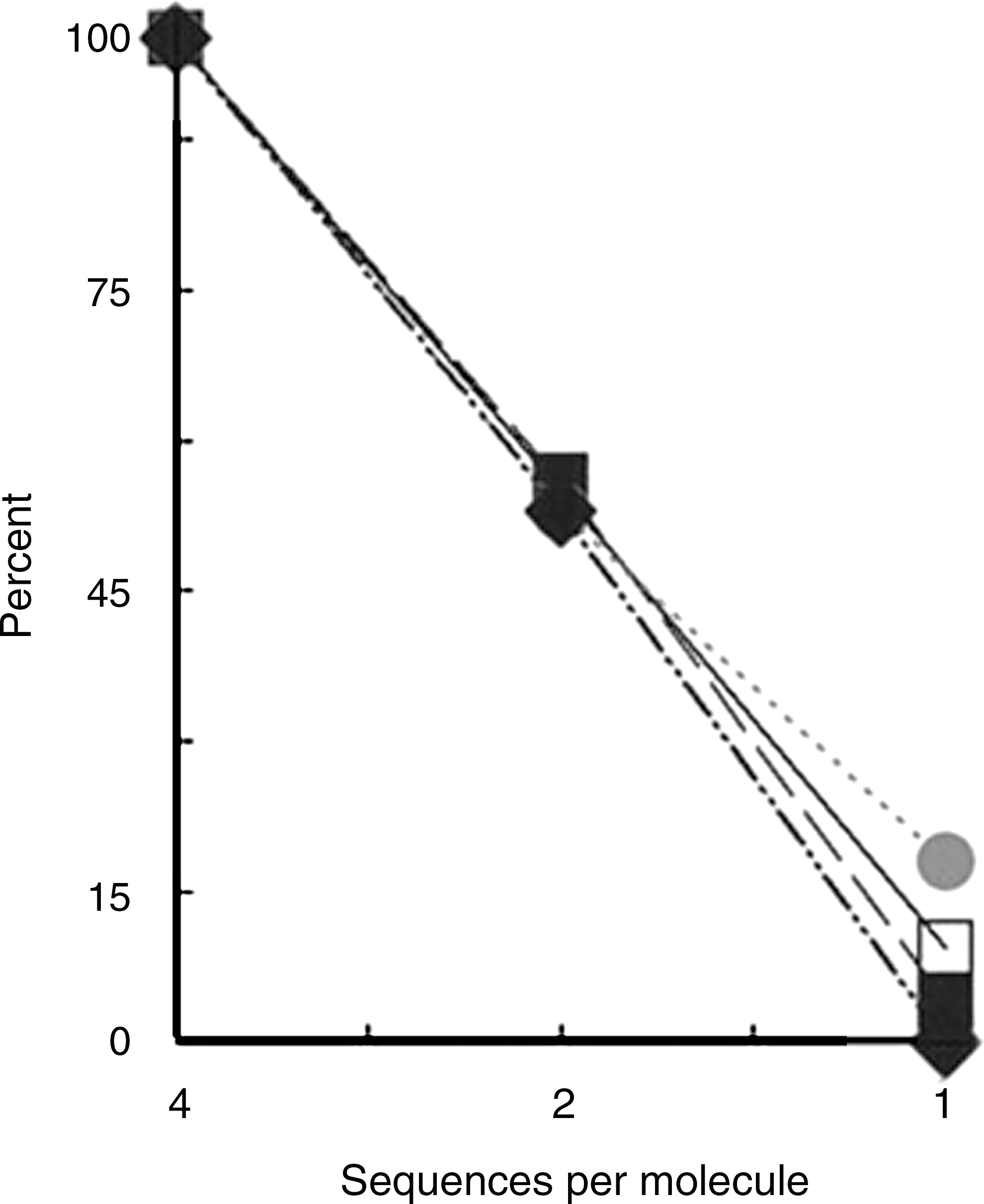
Binding of bivalent or monovalent HPSLK peptides as compared with the quadravalent peptide to lectins. Binding of the quadravalent peptide was set as 100%. The assay wells contained 25 pmol of the quadravalent peptide, 50 pmol of the bivalent peptide, and 100 pmol of the monovalent peptide to provide an equal number of sequences per well. Lectin designations are •, WGA; □, HP; ▪, GS; ♦, DB.
Figure 1 shows that only a low amount of DB, WGA, and GS remained bound to the NPSHPSLG peptide after the extensive washing steps in the binding assay, while binding was not detected with another quadravalent peptide containing the sequence NPSHPLSG, in which the S and L positions were exchanged. We questioned whether the longer peptides would mimic more complex glycans. Lectin SNA1 from Sambucus niger is highly specific for Neu5Ac(α2-6)Gal and does not bind significantly to the monosaccharide, Neu5Ac. 60,61 Binding to lectin MAA from Maackia amurensis requires a trisaccharide and is specific for the Neu5Ac(α2-3)Gal linkage. 61,62 As shown in Fig. 1, the two longer peptides, as well as HPSLK, bound strongly to these two lectins. In these assays, with the lectin bound to the solid phase, the effect of concentration of peptide indicated that half-maximal binding of HPSLK to SNA1 was obtained at 20 nM and to MAA at 400 nM. In contrast, half-maximal binding of NPSHPLSG to SNA1 was obtained at 200 nM and to MAA at 100 nM (data not shown). Thus, although the peptides bind to both lectins, HPSLK bound with higher affinity to the lectin specific for the α2-6 linkage whereas NPSHPLSG had greater affinity for the lectin specific for the α2-3 linkage. These experiments demonstrate that the sequence of the peptide, in addition to valency, is critical for high affinity to these lectins. As an additional control, peptides that lacked the biotin tag were not retained in the assay. We chose HPSLK and NPSHPLSG, based on data shown in Fig. 1, for further studies on activation of phagocytic cells.
Activation of phagocytosis
We determined whether the peptides stimulate uptake of opsonized beads. For these experiments, the HPSLK and NPSHPLSG peptides were synthesized without the C-terminal, ɛ-biotinyl-lysine extension to avoid adventitious binding of the biotin moiety. Antibody-mediated phagocytosis was assayed by the uptake of streptavidin-coated microspheres to which biotinylated HIV-1 envelope gp41 was bound and then opsonized with rabbit antibodies against gp41. PBMC cultures were incubated 20 h with 50 nM peptide and then challenged with the microspheres. As shown in Fig. 3A, an hour after adding the microspheres, cells in untreated cultures contained few if any beads. A relatively inactive quadravalent peptide containing the sequence VSNQH did not significantly stimulate uptake of beads over the vehicle control (Figs. 3B,C). Cells treated with interferon (IFN)-γ as a positive control actively engulfed the microspheres (Fig. 3D). Shown in Figs. 3E–G are macrophage-like cells in cultures treated with the HPSLK peptide, which exhibited spreading and ruffled edges typical of activated macrophages. Similar observations were made with NPSHPLSG (Figs. 3H and I). A quantitative analysis of the number of beads per cell, which shows a highly significant effect of the peptides, is provided in Table 1. In other experiments, streptavidin-coated microspheres, opsonized directly with antistreptavidin antibodies, were engulfed to the same extent as shown in Fig. 3, which demonstrated that the effect of peptides was not specific to the antigen or antibody. Internalization of beads was nearly completely blocked with 50 μM cytochalasin D, an inhibitor of phagocytosis, 63 as assayed by flow cytometry. In these experiments, the inhibition by cytochalasin D of peptide-stimulated uptake of beads was similar to that with cells treated with IFN-γ (data not shown).
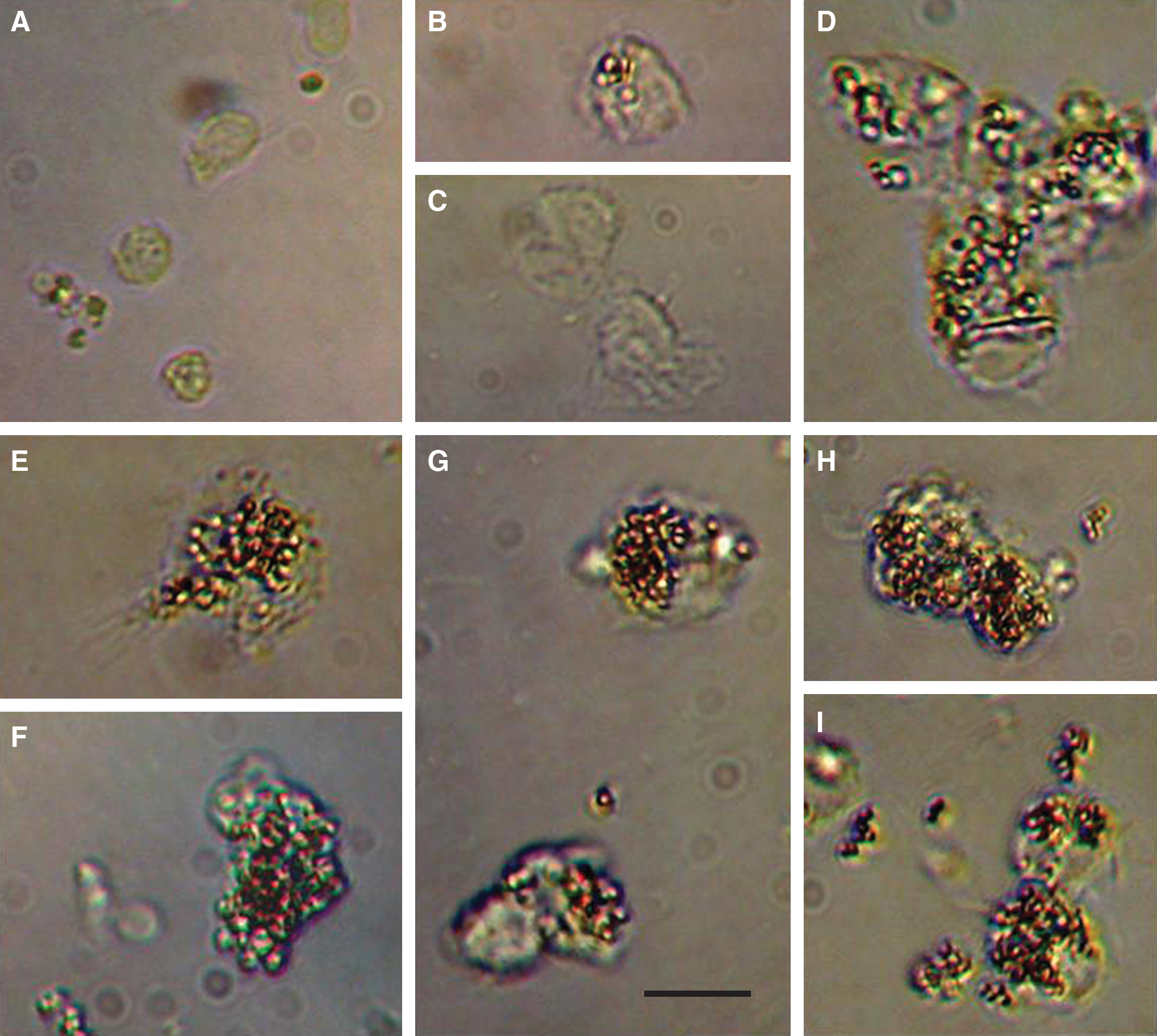
Phagocytosis of opsonized microspheres by peptide-treated PBMCs. Cells were incubated with 50 nM peptides for 20 h, with microspheres for 1 h, and then fixed with 2% formalin as described under Materials and Methods. Free microspheres were removed by washing three times with PBS and the samples were examined by microscopy. (
PBMC cultures were treated 20 h with vehicle (PBS), 50 nM peptide, or 100 ng/ml IFN-γ and then challenged 1 h with microspheres. Cells were fixed with 2% formalin and washed as described in the legend to Fig. 3. Beads in each cell were counted on microscope images, with 15 cells analyzed for each treatment. Uptake of beads in cells treated with peptide VSNQH was not significantly different from the vehicle control (paired Student's t-test, p = 0.702). Paired Student's t-test analyses provided p-values for the other treatments compared with the control peptide VSNQH.
Cytokine response to peptide treatment
Because IFN-γ, a proinflammatory cytokine, stimulated phagocytosis of microspheres, we determined whether the peptides induced release of cytokines. Cultures of PBMCs were treated 4 h with 100 nM HPSLK or NPSHPLSG peptides and media were recovered for analysis of cytokines. With HPSLK, modest increases of 2- to 4-fold, as compared with untreated controls, were found with I-309, IL-17, TNF-β, and TIMP2. A nearly 10-fold increase was found in IL-16. The amounts of these cytokines in the media of cultures treated with NPSHPLSG were not significantly different from the untreated control values. No changes were detected in the release of proinflammatory cytokines such as IL-1α, IL-1β, IL-2, IL-6, IL-8, TNF-α, or IFN-γ with either peptide (data not shown).
Inhibition of HIV-1 replication
IL-16 suppresses HIV-1 replication through inhibition of viral entry, transcription, and desensitization of HIV-1 receptors. 64 –66 Thus we determined whether the peptides inhibit replication of HIV in cultures of PBMCs. Peptide and the virus were added at the beginning of a 3-day incubation period, with peptide at concentrations from 60 pM to 1 μM. At the end of the 3-day incubation, the cells were washed to remove unbound virus, peptide, and antiserum, and again suspended in medium. After an additional 24-h incubation to allow viral replication, each sample was assayed for protein p24 to quantify the amount of virus after cells were lysed by addition of detergent. This assay therefore detected cell-harbored as well as released virus particles.
HIV-1 SF162, an R5, macrophage-tropic strain of clade B that is a major subtype in North America, and HIV-1 97ZA009, an R5 strain of clade C that is a major subtype in Africa, were used as test virus samples. With both subtypes, the HPSLK peptide alone achieved a reduction in replication of the clades B and C strain by 60–80% at concentrations above 100 nM (Fig. 4A, open symbols). Considerable variability in the extent of inhibition was observed, yet 20–40% inhibition was found at concentrations as low as 1 nM. To compare the inhibition of HIV replication by peptide with that provided by antibodies, pooled serum from AIDS patients was diluted into cultures to achieve virion-antibody complexes. These North American patients were presumed to be infected with clade B HIV-1. When the peptide was added along with antiserum at a dilution (1:360), which alone caused only about 30% neutralization, clade B virus expression was reduced to an undetectable level (Fig. 4A, filled squares). The peptide/antiserum combination was nearly as effective against the heterologous clade C strain of the virus (Fig. 4A, filled circles).
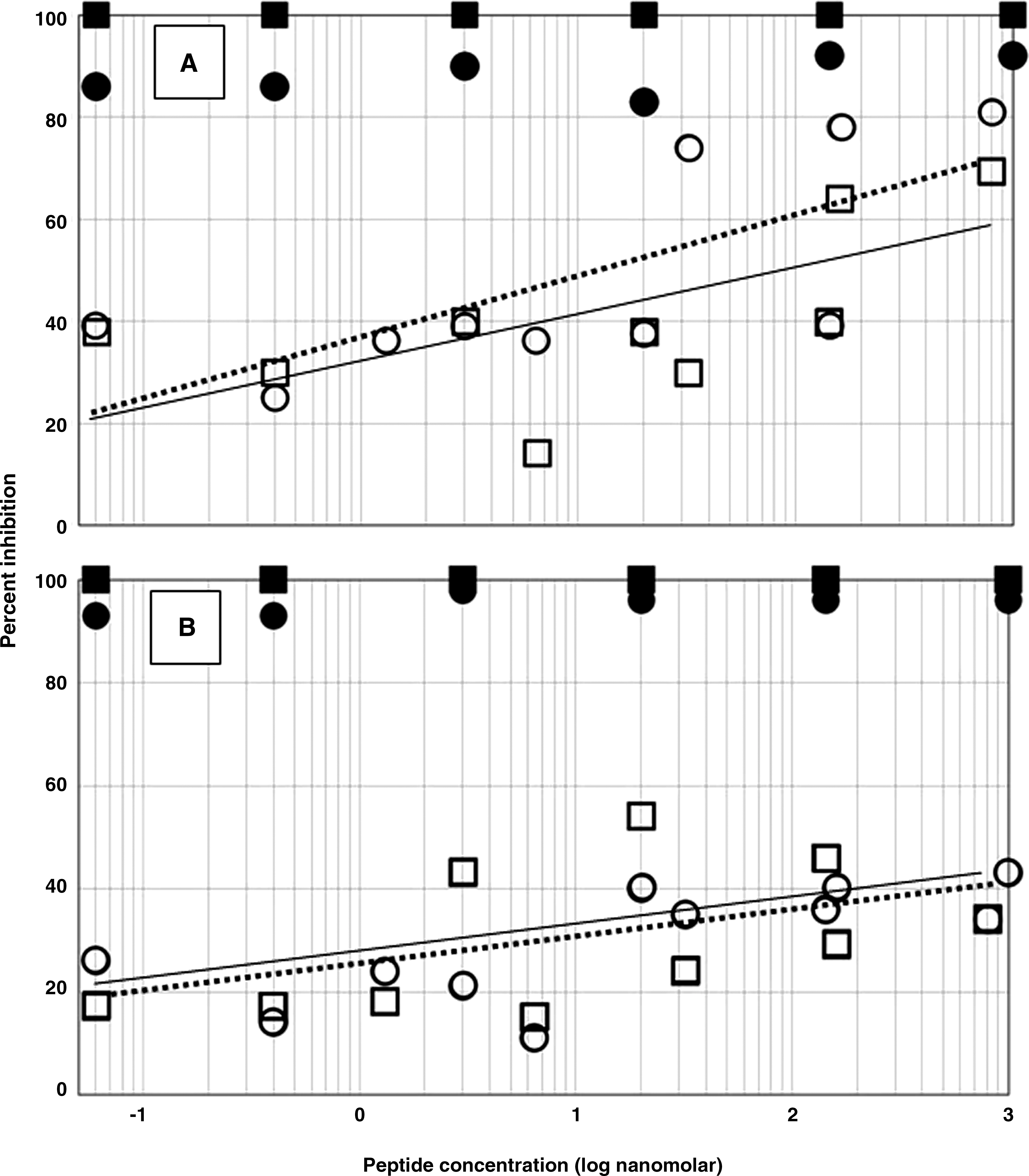
Inhibition of HIV-1 replication in PBMCs cultured with quadravalent peptides at concentrations of 60 pM to 1 μM (note log scale). Open symbols, peptides alone; closed symbols, peptides added with 1:360 dilution of serum from HIV-positive patients infected with a clade B virus. Antiserum alone provided 30% inhibition. (
The effect of the NPSHPLSG peptide on HIV-1 replication was also assayed. As shown in Fig. 4B, results similar to those with HPSLK were obtained, although the inhibition with peptide alone was less. The lower inhibition with the longer peptide alone possibly resulted from the reduced release of cytokines by PBMCs as compared with HPSLK. Nevertheless, in the presence of antiserum the peptide completely inhibited replication of clade B virus and nearly as strongly inhibited replication of the clade C virus. Experiments were also performed with peptide concentrations at 1 pM and lower, i.e., lower than those shown in Fig. 4. At these concentrations, the data were not reliable. Inhibition with peptide alone was not significant and in the presence of antiserum approached that of the antiserum alone (data not shown).
From the data shown in Fig. 3, we reasoned that the presence of phagocytic cells in the PBMC cultures was a factor in the complete inhibition of HIV replication. Therefore, we determined whether the peptides enhanced inhibition of HIV replication by antiserum in MT2 cells, a T cell line that lacks phagocytic activity. Addition of undiluted antiserum completely neutralized the virus. At a dilution of 1:71, the antiserum alone inhibited viral replication by 35%. Addition of peptide HPSLK to a final concentration of 10 nM or 1 μM did not increase inhibition (Fig. 5). Similar results were obtained with peptide NPSHPLSG. For comparison, the same experiment was performed with PBMCs. A greater dilution (1:360) of the antiserum was required to reduce neutralization to 30%. However, addition of peptide HPSLK caused complete inhibition of viral replication (Fig. 5). We attribute the lack of effect of a peptide in with MT2 cells to the absence of antibody-mediated phagocytic activity.
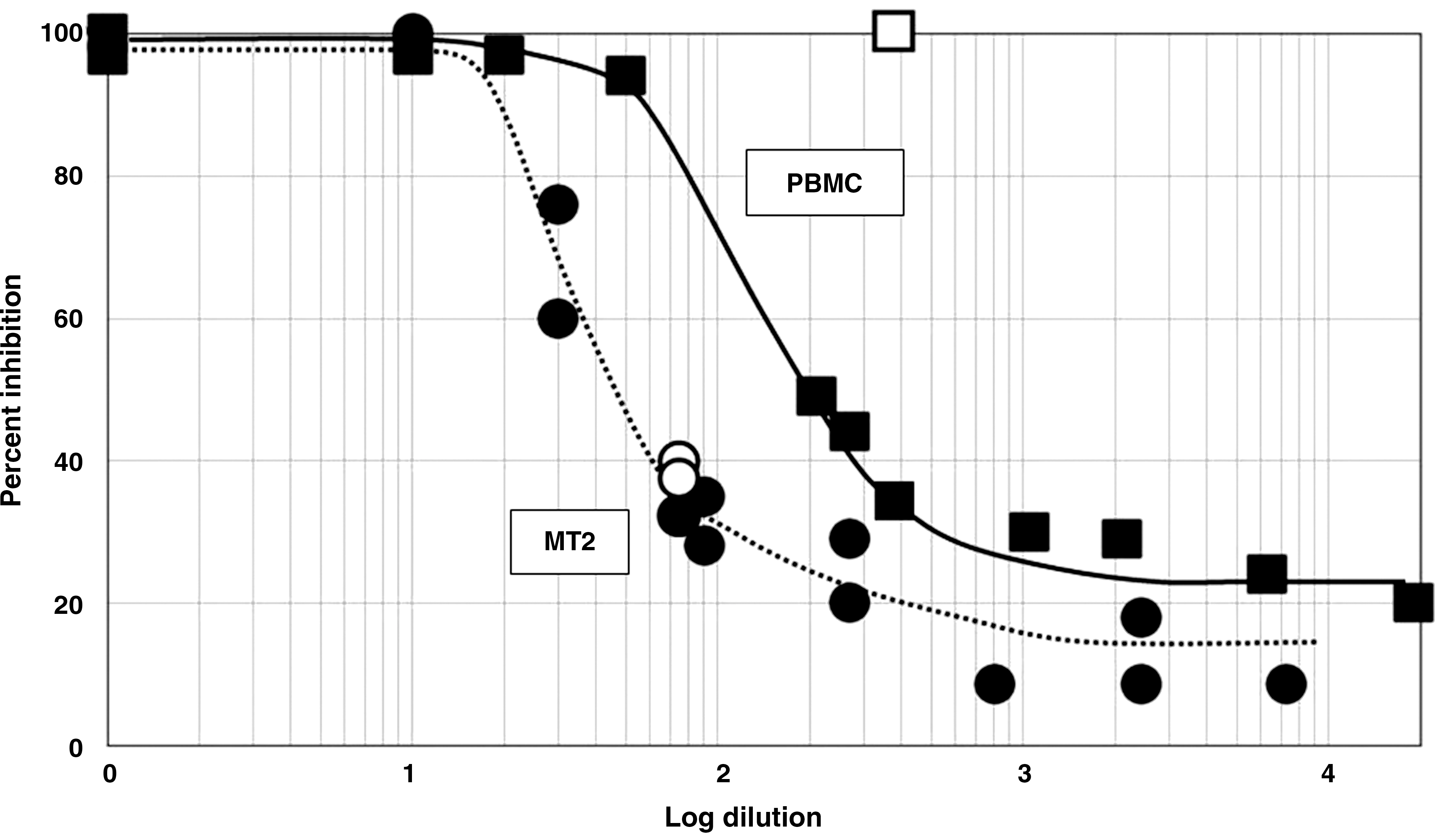
Inhibition of replication of HIV-1 (•) strain 23135 in MT2 cells or (▪) strain SF162 in PBMCs containing a dilution series of pooled sera from HIV-positive patients. Each curve is the combined results of two separate experiments. Dilutions of serum that provided about 30% inhibition were chosen to test the effect of added HPSLK peptide. The two open circles near the curve for MT2 cells indicate results with 10 nM (lower symbol) or 1 μM (upper symbol) peptide. Inhibition was 100% with peptide concentrations between 10 nM and 1 μM in PBMC cultures (open square) (see Fig. 4).
Cytotoxicity assays
To ensure that the peptides did not prevent viral replication by impairing the cells, their effect on cell viability was determined. The experimental conditions were similar to those for Fig. 4 but did not contain virus. At concentrations of peptide of 1 μM or less, the number of viable cells was no less with peptide as compared with the vehicle, whether the vehicle was PBS or antiserum diluted in PBS (1:360) (Fig. 6). The HPSLK peptide reduced viability to about 50% at a concentration of 1 mM, which is 106-fold greater than a concentration that was effective in inhibition of HIV replication. Viability was slightly increased at all concentrations of the NPSHPLSG peptide.
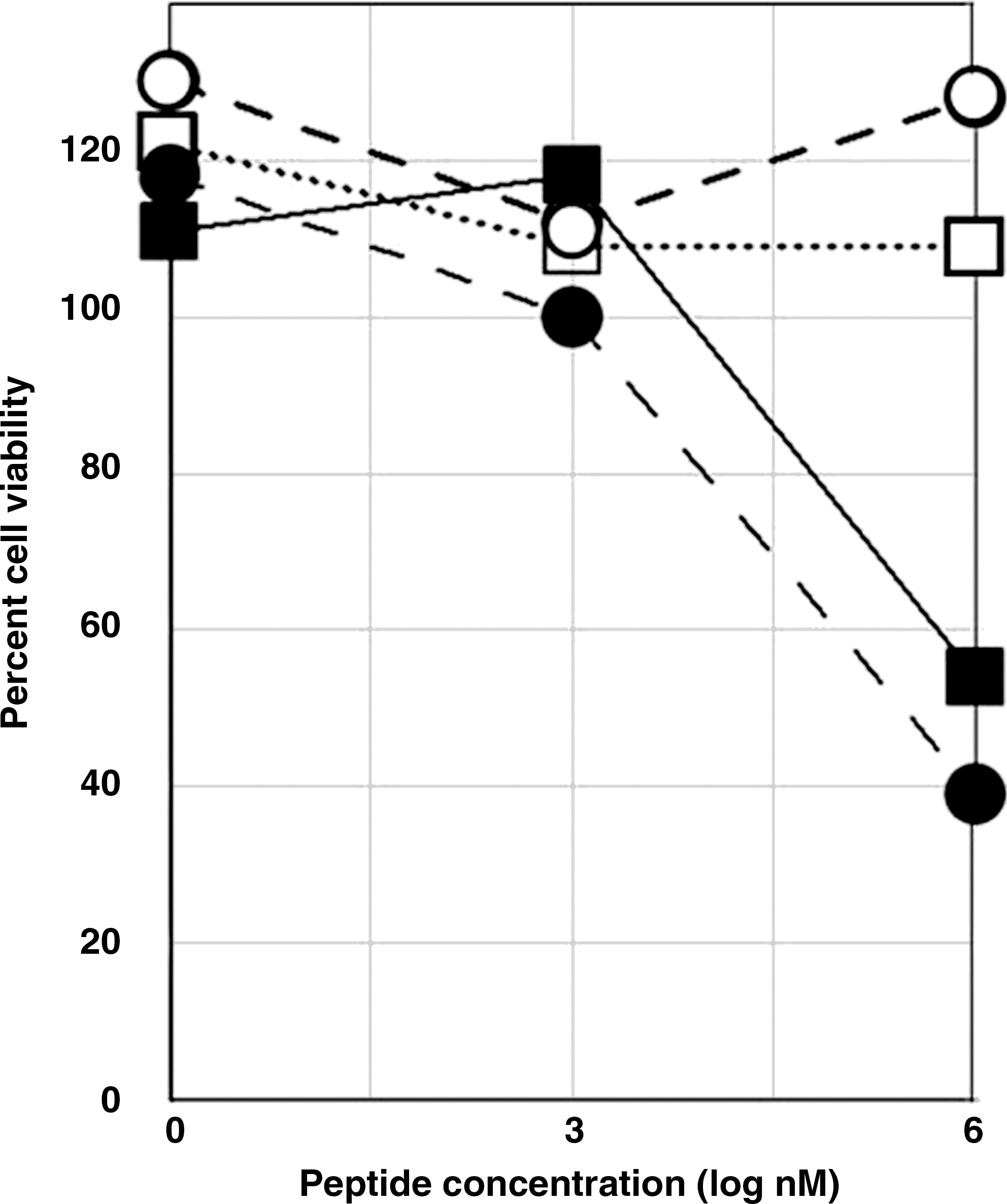
Cytotoxicity of quadravalent HPSLK (closed symbols) or NPSHPLSG (open symbols) in PBMC cultures. Viability with peptide was compared with vehicle alone, either PBS (circles) or a 1:360 dilution of HIV-positive serum in PBS (squares).
Discussion
As demonstrated in Fig. 2, quadravalent peptides bound lectins approximately two times more strongly than divalent peptides and 10 times more strongly than monovalent peptides. These differences in binding affinity, with equal numbers of sequences, were predicted from entropic factors 54,55 and ligand density. 56 A multivalent peptide is also essential to facilitate cross-linking of receptors, which is often required for transduction of a signal from the cell surface. 36 When synthesized as a quadravalent molecule, with four branches extended from a trilysine core, the HPSLK peptide bound with high affinity to several lectins with differing monosaccharide specificities. Under the conditions of these assays, peptide HPSLK had characteristics of a general sugar mimetic. Competition assays indicated that the K d value for binding to WGA was about 7 nM for the quadravalent peptide. Because only lectin that was retained after extensive washing in the assay was measured, the actual affinity in solution may be even greater. The K d values can be considered as only approximations of binding affinities to cellular receptors. Further support for the peptide interacting with the sugar-binding site of the lectins was the strong inhibition of binding to the lectins by mucin, which contains a variety of complex glycans that bind more strongly than single sugars.
Peptides with a longer sequence, such as NPSHPLSG, exhibited more restrictive binding. This peptide did not bind strongly to lectins that bind monosaccharides but bound with high avidity to those that bind complex glycans. NPSHPLSG bound more strongly to lectin MAA, which is specific for the trisaccharide Neu5Ac(α2-3)Gal(β1-4)GlcNAc 62 than to SNA1, whereas HPSLK bound more strongly to SNA1, which is specific for the disaccharide Neu5Ac(α2-6)Gal. 60,61 SNA1 and MAA do not bind the monosaccharide, Neu5Ac. We found that half-maximal binding of the peptides to these lectins was in the low nanomolar range. Shibuya et al. 60 and Knibbs et al. 62 found in precipitation reactions that these lectins were inhibited 50% by 12 μM Neu5Ac(α2-6)lactose or 500 μM Neu5Ac(α2-3)lactose, respectively. Thus the affinities of the peptides to these lectins are roughly 103-fold greater than binding of oligosaccharides and 104-fold greater than that of monosaccharides. 67 The affinity with which the NPSHPLSG peptide binds SNA1 and MAA, considering the extensive washing steps in the assay, was similar to binding of these lectins to solid-phase glycan arrays. 61
These features suggest that the longer peptides favorably mimic complex glycans such as ligands for a variety of siglecs, which also do not bind significantly to a single Neu5NAc. 19,20 Most siglecs are on cells of the immune system, contain an ITIM in their cytoplasmic domain and are considered to be inhibitory receptors. 18,19 Analogous to the binding of peptides to lectins, Carlin et al. 68 showed that an amino acid sequence within the N-terminal domain of β protein of group B Streptococcus binds to siglec-5, which contains an ITIM in its cytoplasmic domain, and inhibits phagocytosis of the bacterial cells. In contrast, siglec-14 and siglec-15 have short cytoplasmic tails that lack ITIMs and serve as activating receptors in concert with adaptor proteins such as DAP12, which contains an ITAM in its cytoplasmic domain. 24,25 Monocytes, macrophages, and dendritic cells also have many C-type lectins on their surface that are activating receptors. 15 The possibility exists that the peptides interact with one or more of these receptors to stimulate phagocytic activity.
Interestingly, NPSHPLSG appears to be less cytotoxic than HPSLK, which may result from its more restrictive binding characteristics. We did not find a significant increase in cytokine release in PBMC cultures in response to treatment with the longer peptide. However, both peptides were as active in stimulating phagocytosis of microspheres coated with anti-HIV antibody as IFN-γ (Fig. 3), a highly inflammatory cytokine. 69 The NPSHPLSG peptide in particular may stimulate phagocytosis without inducing release of proinflammatory cytokines. Whereas human anti-HIV antibodies were used in the HIV replication assays, microspheres were opsonized with rabbit anti-gp41 or antistreptavidin antibodies, which indicated that the stimulation of Fc-mediated phagocytosis was not antigen or antibody specific.
Of particular interest was the complete inhibition of replication of the clade B strain of HIV-1 in PBMCs in the presence of pooled antisera obtained from HIV-1 clade B-infected patients, which alone neutralized only 30% at the dilution used in these assays. The antiserum appears to contain nonneutralizing antibodies that nevertheless are capable of virion clearance when phagocytes are stimulated by the peptides. Insufficient virus remained at the end of the 3-day treatment with peptide to provide a detectable amount of protein p24 from clade B virus.
The peptides inhibited replication of HIV-1 over a wide concentration range in these cultures, with inhibition nearly as strong at 1 nM as at 1 μM, which was indicated by the shallow slopes of the regression curves for peptide alone in Fig. 4. Possible explanations for this effect are, first, that given the K d of binding to lectins, receptors may have been saturated at the higher concentrations of the peptide. Second, each quadravalent molecule carries a high local concentration of the peptide, and thus its effects are expected to be less dependent on stochastic interactions of multiple molecules. As clustering of receptors is required for many cellular effects, 19,36 each multivalent molecule alone can apparently serve this purpose as the result of ligand density. 56
Acute HIV infection elicits both neutralizing antibodies that block fusion of the virus with host cells and nonneutralizing antibodies, 70 as reflected in the titration of pooled antisera shown in Fig. 5. Antibodies bind the virus and provide the opportunity for phagocytic cells to destroy virion-antibody complexes (“opsonized” virus) by Fc-Fcγ receptor (FcγR)-mediated phagocytosis. A number of research groups found that viral replication was attenuated by interaction of such complexes with FcγR on macrophages and immature dendritic cells 4 –9 and that activation of macrophages in vitro reduced viral load in PBMC cultures. 8 Hessell et al. 9 showed that interaction of the Fc portion of antibodies with receptors (FcγR) on phagocytes is important in reducing viral yield from infected cells and, moreover, that the Fc-FcγR interaction is more important than the complement system. Nevertheless, concern has been expressed that binding of complement to antibody-complexed virions may counteract the immune system and cause complement-dependent, antibody-dependent enhancement of infection. 71 Based on the lack of detectable p24 at the end of the incubation period, shown in Fig. 4, our data are consistent with the suggestion 4,5 that virion-antibody immune complexes, formed even with nonneutralizing antibodies, enter a degradative pathway when phagocytic cells are activated by the peptides. Future studies will examine in detail the mechanism of this effect.
Our results suggest that the peptides mimic complex glycan structures and stimulate antibody-mediated phagocytosis. This activity may be a factor in providing complete inhibition of HIV-1 replication in vitro. Further studies are needed to confirm phagocytosis as the mechanism of the extraordinary synergism of the peptides and antibodies. Administration of very low concentrations of the sugar-mimetic peptides alone may have therapeutic potential in AIDS patients through synergy with autologous HIV antibodies.
Note Added in Proof
Further characterization of the peptides described in this report is presented in Eggink LL and Hoober JK: Peptide mimetics of terminal sugars of complex glycans. Glycobiology Insight 2010:2. (This is a new electronic journal.)
Footnotes
Acknowledgments
L.L.E. and J.K.H. designed the peptides and performed the lectin binding assays and phagocytosis assays. M.S. and C.V.H. performed the HIV proliferation assays. J.K.H. drafted the manuscript and all authors contributed to its editing. We gratefully acknowledge Daniel Brune and John Lopez, Proteomics and Protein Chemistry Laboratory, Arizona State University, for peptide synthesis.
Author Disclosure Statement
L.L.E. and J.K.H. declare that they are inventors of technology contained in this report. Intellectual property has been assigned to Susavion Biosciences, Inc. in which the inventors hold shares.