
Abstract
In this study, we characterized four HIV-1 strains from Cameroon, Gabon, and the Democratic Republic of Congo (DRC), collected during independent serosurveys, and previously found to cluster in the pol gene with HIV-1 MAL and HIV-1 NOGIL3, two complex recombinant viruses reported in the early HIV epidemic, and with the recombinant strain 04FR.AUK recently described in France. The four newly sequenced viruses shared the same structure as 04FR.AUK, involving alternating fragments of subtype A, K, and unclassified (U) fragments, representing a new CRF called CRF45_AKU. Some of the unclassified fragments were related to unclassified regions described in either CRF04 or CRF09 strains. Careful reanalysis of HIV-1 MAL and HIV-1 NOGIL3 demonstrated that these strains were related exclusively to CRF45_AKU and either two subtype D fragments for HIV-1 MAL or one subtype H segment for HIV-1 NOGIL3. Following extensive blast searches, related gag, pol, and env sequences were observed in Central and West Africa (Senegal, Mali), as well as in Europe (France, Spain, Italy, Cyprus), Argentina, and China.
H
The global distribution of HIV-1 isolates is heterogeneous around the world and is a dynamic process. 3,4 Although the epidemic in the western world is primarily due to subtype B, the current global HIV-1 epidemic is dominated by non-B variants. The highest genetic diversity is observed in Africa, particularly in Central Africa where all HIV-1 groups and subtypes and high numbers of CRFs and URFs cocirculate. In addition, at least 5% of HIV-1 strains cannot be classified. 5,6 The genetic distance between the two oldest HIV-1 samples, DRC60 and ZR59 from Kinshasa, the capital city of the Democratic Republic of Congo (DRC), directly demonstrates that diversification of HIV-1 in West-Central Africa occurred long before the recognized AIDS pandemic and provides evidence that the HIV-1 group M pandemic most likely originated in this geographic region. 5,7
Recently, we described the near-full-length sequence of a complex recombinant HIV-1 strain, 04FR.AUK, involving subtypes A, K, and unclassified (U) fragments in a patient originating from the DRC but whose primary infection occurred in France. 8 Interestingly, in the gag-pol region this new virus clustered with some of the oldest recombinant strains, HIV-1_MAL and HIV-1_NOGIL3, from DRC and Central Africa. 9,10 We also identified viruses that based on partial pol and/or gag and/or env sequences were closely related to the 04FR.AUK strain. In this study we report the full-length genome sequences of four HIV-1 strains from Central Africa sharing the same complex recombinant structure involving subtypes A, K, and unclassified fragments, as 04FR.AUK, which thus represent a new CRF: CRF45_AKU. The strains were isolated in 1997 during independent serosurveys conducted in several countries from West and Central Africa: 11 –13 DRC (97CD.MBFE185 and 97CD.MBS30), Gabon (97GA.TB45), and Cameroon (97CM.MP814).
The HIV-1 strains characterized in this study were amplified from uncultured patient peripheral blood mononuclear cells. DNA was extracted using the Qiagen (Qiagen SA, France) DNA isolation kit. Complete sequences were obtained by amplifying large overlapping fragments, using different sets of primers previously reported for HIV-1 group M. 8 Due to the length of the targeted fragments, we used the Taq Expand Long Template PCR as previously described. 14 The amplified fragments were purified using a QiaQuik gel extraction kit (Qiagen, France), and then directly sequenced using a BigDye Terminator sequencing kit (Applied Biosystems, France). Electrophoresis and data collection were done on an Applied Biosystems 3130XL Genetic Analyzer. The sequenced fragments from both strands were reconstituted using Seqman II from the DNAstar package v5.08 (Lasergene, USA). Four nearly complete genomes were obtained; all sequences were lacking two-third of the 5′-LTR: 9086 base pairs (bp) for 97GA.TB45, 9146 for 97CM.MP814, 9158 for 97CDMBFE185, and finally 8536 for 97CD.MBS30, which was not amplified in the 3′-LTR region. For all strains the reading frames were open and of complete length.
The new sequences were aligned with reference sequences using Clustal X and were compared with all pure subtypes including subsubtypes (A1–A4, F1/F2), CRFs circulating in Central Africa, or constituted with subtypes circulating in those regions (CRF01, 02, 04, 05, 06, 09, 11, 12, 13, 14, 18, 19, 20, 22, 23, 24, 25, 26, 27, 36, 37, 43). We also included 04FR.AUK,
8
two unclassified sequences, tentatively called subtype L but designated as UL (AF286236, AF457101), one sequence (95NL-H10986/EF029066) close to subtype K, and also the old recombinant strains HIV-1_MAL and HIV-1_NOGIL3.
9,10
Phylogenetic analyses of the near-full-length sequences were done using the neighbor-joining (NJ) method and 1000 bootstrap replicates to estimate the reliability of the branching orders as implemented in CLUSTAL X. FigTree version 1.2.1 (Andrew Rambaut, University of Edinburgh,
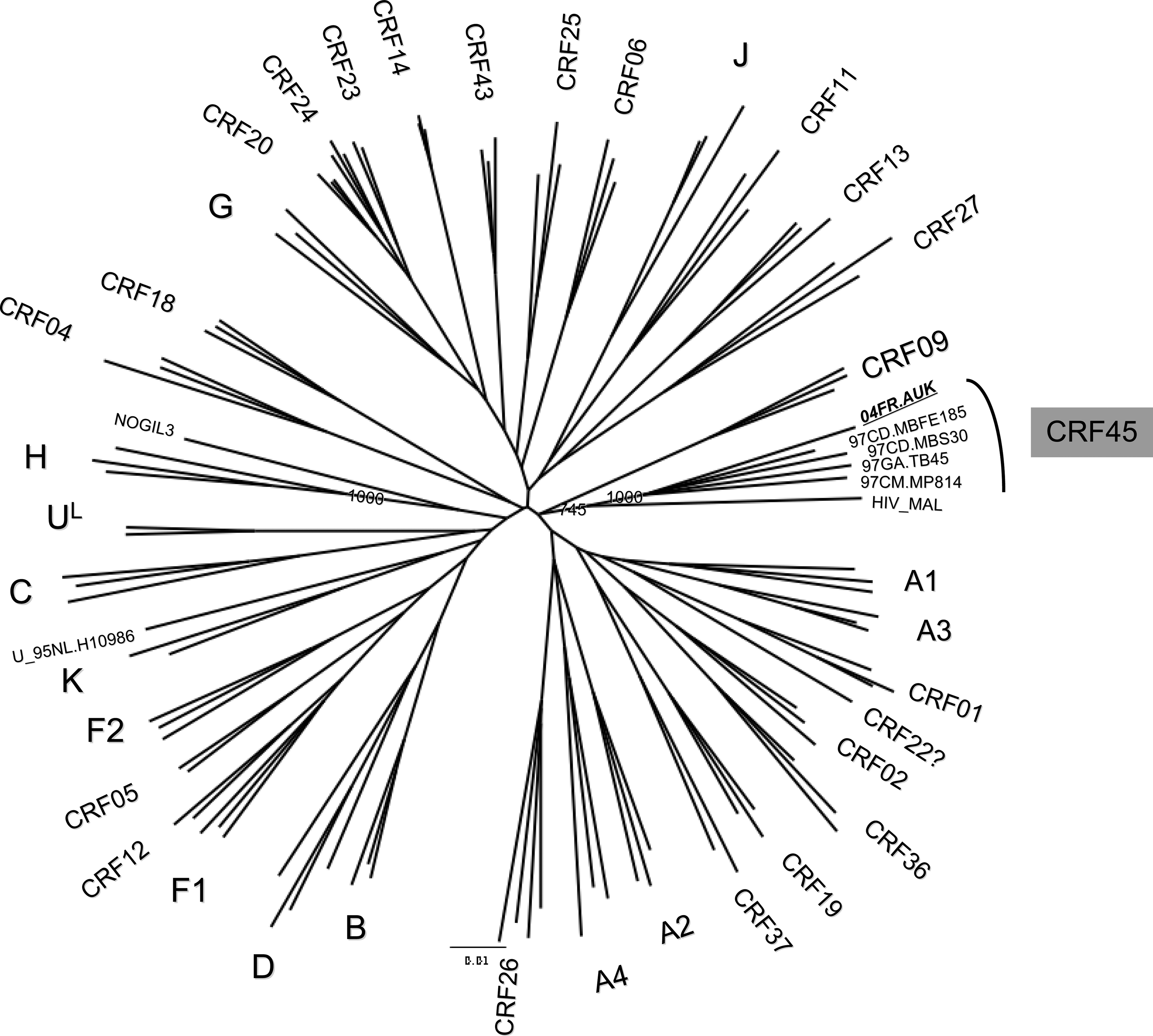
Phylogenetic relationships of the near-full-length genomes representing the overall HIV-1 genetic diversity with the newly sequenced strains. The phylogenetic analysis was done using the NJ method with 1000 bootstrap resamplings on 7771 unambiguously aligned nucleotides. Only the bootstrap values at the node of the newly sequenced strains were indicated; those supporting the other clades or subclades were all 1000.
To determine whether each of the four new sequences shared the same mosaic structure as 04FR.AUK, we performed recombination analyses on the same alignment of HIV-1 reference sequences as used in the phylogenetic analysis. The Simplot v2.5.1 software (Stuart Ray,
In a first step, a similarity analysis was conducted for each new sequence plotted against all reference strains and the prototype 04FR.AUK. Each new strain was closely related to 04FR.AUK along the entire genome and not to any other HIV-1 variant (results not shown). In a second step, bootscan analysis was done for each new sequence plotted against the reference strains for the pure subtypes only. We observed similar mosaic patterns for all strains with alternating fragments of subtypes A and K and four small segments that did not fall into the actual known subtypes/CRFs (noted U). The left panel of Fig. 2 illustrates the bootscan plots obtained for the new strains compared to 04FR.AUK. In a third step, we added separately all other known HIV-1 variants including subsubtypes and CRFs in order to examine the unclassified fragments in more detail. These additional analyses also showed that some of the unclassified fragments were closely related to unclassified fragments observed in pol for CRF04cpx and in env for CRF09cpx.

Recombination analysis of each of the newly sequenced strains and the previously reported 04FR.AUK strain compared to the pure HIV-1 subtypes. The bootscan analysis implemented with Simplot 3.5.1 beta software was done on sliding 450 nucleotide windows with 10 nucleotide increments (left panel). The full-length genome alignment was divided into 12 independent segments corresponding to different subtype designations by bootscan analysis. After gap-stripping, each segment was submitted to NJ phylogenetic tree analysis with 100 bootstrap resamplings (right panel). The bootstrap values are indicated only for the cluster of the newly sequenced strains. Those supporting other clusters were higher than 70. The new strains are indicated only with the sample code for better clarity and are in bold. Each region was numbered, and the subtypes of the new strains were indicated as well as the length of the corresponding gap-stripped alignment.
We then confirmed by phylogenetic tree analysis the different segments observed in the bootscan plots. The phylogenetic trees were constructed with the minimal number of references, i.e., with pure subtypes and excluding CRFs that were not present in the mosaic pattern of the newly obtained sequences. The bootscan analyses defined 12 different segments as illustrated in the left panel of Fig. 2. The corresponding phylogenetic trees are shown in the right panel of Fig. 2. The lengths of the segments are given as unambiguously aligned nucleotides from gap-stripped alignments. The HIV-1_MAL and HIV-1_NOGIL strains were included in the phylogenetic analysis, except in the segments where they are recombinant; more precisely, HIV-1 MAL was omitted from the analysis in gag because the strain recombined with subtype D in that region (see below).
Segment I (1519 bp), encompassing part of the 5′-LTR, the gag gene, and the nearly complete protease region in the pol gene, was clearly related to subtype A. This A subcluster included the CRF09_cpx strains, which are subtype A in this region, and seemed perfectly monophyletic with the new sequences, supported with 90% bootstrap value. Segment 2 (535 bp), located in the beginning of the reverse transcriptase (RT) in pol, was significantly divergent from the actual known subtypes or recombinants. As a result, the subtype/CRF for this fragment could not be resolved and it was designated U, as were the CRF09_cpx strains, but the unclassified segments from the two CRFs were clearly different from each other. Segment 3 (390 bp), also corresponding to a fragment of RT, was clearly subtype K (84% bootstrap value) and included again the CRF09_cpx strains, which are also K in this region. Segment 4 (260 bp) was unresolved in the bootscan plot of the prototypal 04FR.AUK and in one of the new sequences (97CD.MBFE185); however, it seemed to be subtype K in the three other bootscan plots and was close to the K references in the corresponding tree. Because the bootstrap value supporting the subtype K cluster was low, this segment should be classified as U linked to K and might correspond to a region in which the variability of these strains is higher compared to the rest of the subtype K region. In this fragment, the CRF09_cpx strains were not linked to the new HIV-1 variant and were subtype A. Segment 5 (496 bp), located at the end of the RT, was assigned as subtype K with a 96% bootstrap value. Segment 6 was a very small region of 219 bp that clusters with a U fragment found in the mosaic structure of the CRF04_cpx strains (86% bootstrap value). Segment 7 (1734 bp), spanning the integrase, the accessory genes vif and vpr, the first exons of tat and rev, and one-third of the vpu gene, clustered inside subtype A with 100% bootstrap value. Segment 8 (261 bp), corresponding to the second part of the vpu gene and the beginning of env, clustered again with subtype K (78% bootstrap value). Segment 9 (1960 bp), from the beginning of gp120 to the beginning of gp41, was found to branch inside subtype A as a separate subcluster (89% bootstrap value), and was different from the subtype A fragment from the CRF09_cpx strains. Segment 10 (350 bp) in gp41 was unclassified, but corresponded to a U segment found in the mosaic structure of the CRF09_cpx strains, again perfectly monophyletic with the new sequences (79% bootstrap value). Segment 11 (335 bp), from the end of gp41 from the second part of nef until the major part of 3′-LTR, could not be classified into any known subtype/CRF and was designated U.
The mosaic pattern of the new sequences corresponds to the previously reported recombinant structure of the 04FR.AUK strain, 8 except for the additional segment (12) for the 3′-LTRs that was designated as U and that was not studied for 04FR.AUK. The alteration of subtypes A, K, and the unclassified fragments, as well as the previously observed clustering with the complex CRF09_cpx strains in some segments, was perfectly concordant. Moreover, an unclassified fragment (segment 6) could be better defined due to the existence of five strains instead of one and could be linked to a U fragment present in the CRF04_cpx strains.
Overall, the results indicated that these epidemiologically unlinked viruses shared the same mosaic structure with alternating fragments of subtypes A, K, and U, allowing us to define a new CRF that has been named CRF45_AKU in accordance with the Los Alamos database. Figure 3 shows the schematic representation of the mosaic genome structure of the new CRF, drawn with the Los Alamos database recombinant mapper drawing tool (

Schematic representation of the mosaic genome structure of the recombinant strains drawn with
Because the subtype A fragments from the new strains formed a separate cluster within the subtype A radiation, we performed a SUDI analysis to determine whether these fragments are representatives of a new potential subsubtype within A (
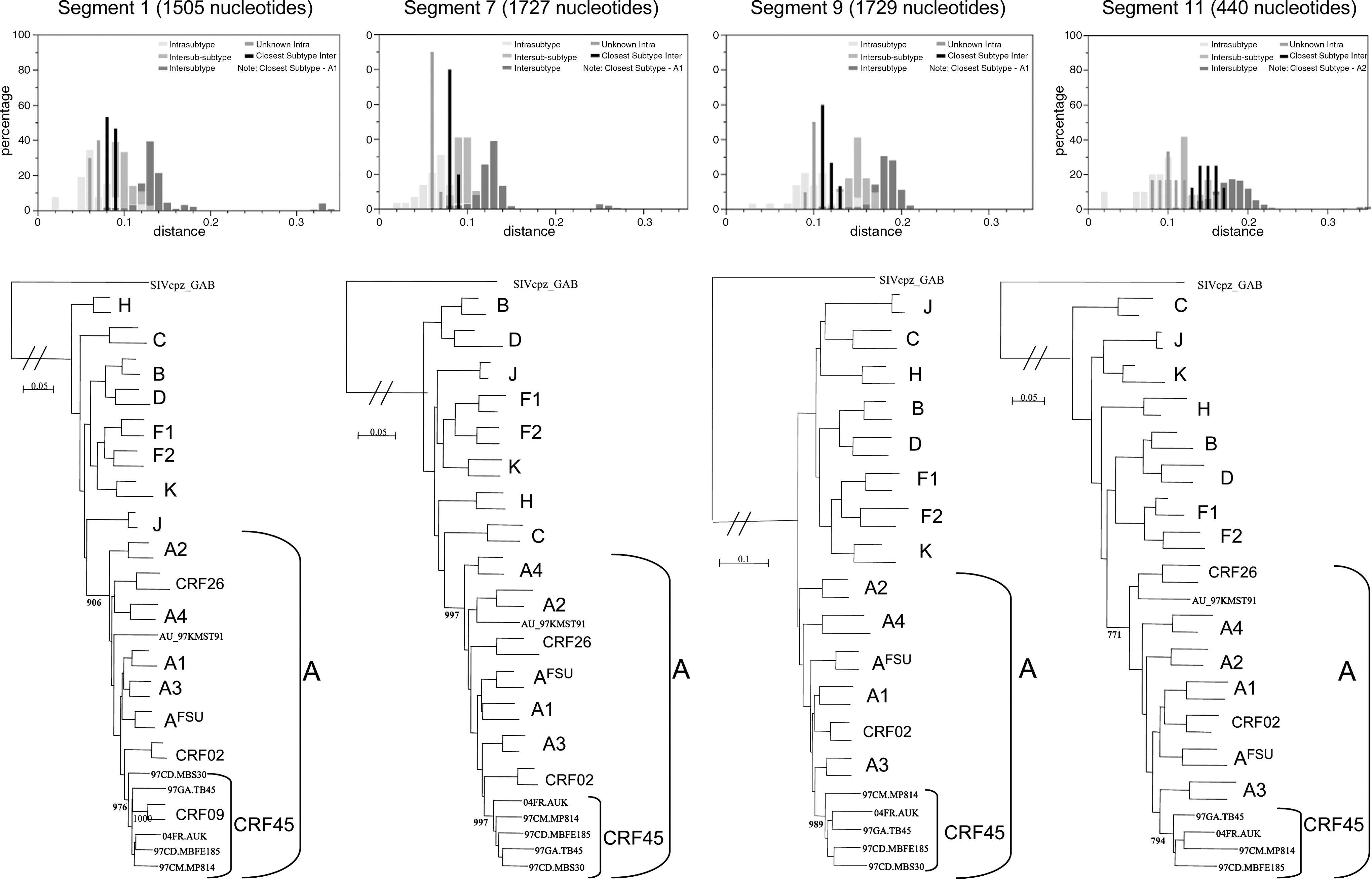
Subtype distance tool output for the A-related segments of the CRF45_AKU strains (top), as defined by the bootscan analysis from Fig. 2. The corresponding maximum likelihood phylogenetic trees (bottom) were constructed from alignments containing SIVcpz_GAB as an outgroup and two reference sequences for each HIV-1 subtype/CRF (A1: KE.Q2317, 92UG037; A2: 94CY017, 97CD.KTB48; A3: DDJ369, DDI579; A4: 97CD.KCC2, 97CD.KTB13; A-FSU: 02IT.60000, 03RU20; A-CRF02: NG.IBNG, DJ.DJ263; CRF09_cpx: 95SN179, 96GH291; CRF26_A5U: 97CD.KTB119, 02CD.LBTB084; unique A from DRC: AU_97CD.KMST91; B: HXB2, US.WEAU160; C: ETH2220, 96BW0502; D: ZR.NDK, 99TC.MN011; F1: 93BR020, FIN9363; F2: CM53657, 96CM.MP255; H: 90CF056, BE.VI991; J: SE91733, SE92809; K: 96CM.MP535, 97CD.EQTB11). The bootstrap values were indicated only at the nodes of the A radiation and for the CRF45_AKU cluster; in all trees, the bootstrap values supporting the different HIV-1 lineages were 817 or higher. The unique 97CD.KMST91 strain and CRF26_A5U have been shown to contain small undetermined subtype segments along the genome, particularly in the gp120, and were therefore excluded from the analysis of region 9.
We also calculated the intraclade genetic distances for each of the A segments of the four CRF45_AKU strains. Table 1 indicates that they corresponded at least to the maximal values for all other intraclade distances, strongly suggesting that the CRF45_AKU variant evolved over a long period of time.
Compared to intrasubtype genetic distances of the other HIV-1 strains.
It was noted that HIV-1_MAL and HIV-1_NOGIL, complex recombinant strains isolated early in the HIV-1/M epidemic, strongly clustered with CRF45_AKU in the 5′-part of the genome. Therefore we reanalyzed the previously reported recombinant structure of these viruses by including the new CRF in the Simplot and bootscan analysis. The results are depicted in Fig. 5a for HIV-1_MAL and Fig. 5b for HIV-1_NOGIL3. The top of the figures shows a similarity plot of the query sequence against the pure HIV-1 subtypes including subtypes A and K and CRF45_AKU; below is the corresponding bootscan plot without subtypes A and K, and finally the schematic representation of the genomic structure. The similarity plots demonstrated clearly that each strain was composed of CRF45_AKU and either subtype D or H exclusively. The bootscan plots were drawn without subtypes A and K in order to avoid competition with the CRF45_AKU signal in this analysis. Therefore HIV-1_NOGIL3 was probably the result of one recombination event between CRF45_AKU and the pure subtype H, while HIV-1_MAL seemed to be more complex since two subtype D fragments are present inside a CRF45_AKU structure or vice versa. These results are perfectly concordant with published reanalysis of intersubtype recombinant sequences comprising HIV-1_MAL and HIV-1_NOGIL3. 17 At the time of this study, the CRF09_cpx sequences were not available and so were not included in the reanalysis.
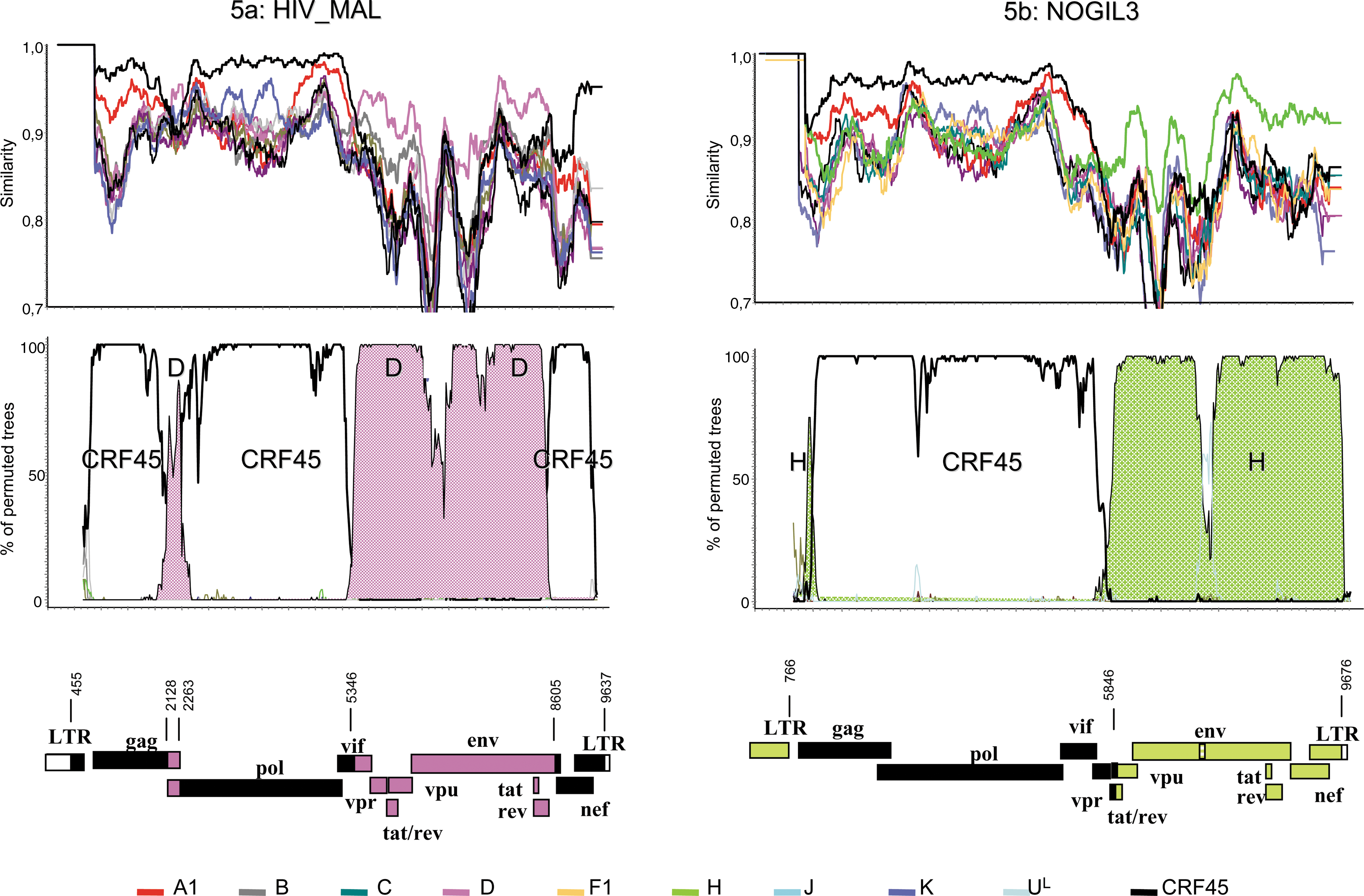
Recombination analysis and schematic representation of the mosaic structures of HIV-1_MAL (
We already reported in the study that described the prototype CRF45_AKU strain 04FR.AUK that we identified viruses with similar structure in gag and/or in pol in Central Africa as well as in Senegal. 8 We here extended these blast searches and identified related sequences in pol in HIV-1 strains described in Europe (France, Spain, Italy, and Norway for the NOGIL strains), South America (Argentina), and Asia (Shanghai, a major city in China). 18 –21 All these sequences were individually checked by recombination analysis and were found to be perfectly concordant with the CRF45_AKU recombinant structure over their available length in pol. Figure 6 shows the NJ tree of those pol sequences from the global gap-stripped alignment, covering part of the RT (565 nucleotides), where they formed a unique cluster with the new strains supported by 75% bootstrap value. However, despite their similar structure in pol, it cannot be excluded that in env these strains are different from 04FR.AUK, like HIV-MAL or NOGIL.

NJ tree of strains from different geographic origins, clustering with the newly characterized CRF45_AUK in the reverse transcriptase region (565 bp). The bootstrap value at the node of the new cluster, indicated by an arrow, was 75. Asterisks correspond to bootstrap values higher than 70 for the other clades and subclades. The reference strains for CRF45_AKU are highlighted in pink and HIV-1_MAL and HIV-1_NOGIL3 are in green. Each strain clustering inside the newly described lineage is indicated with its accession number when available, and a line is drawn to its country of isolation on the world map.
Interestingly, one strain collected in the mid-1980s in Kinshasa (DRC) 22 was clearly inside the CRF45_AKU cluster, but it was not included in the tree because only the protease sequence was available. Some other available pol sequences were excluded from the tree because they were not exactly concordant with the CRF45_AKU mosaic structure. This was the case for several samples from Gabon previously described as CRF MAL-like. 23
We also found that two strains (CY111 and CY112, accession numbers FJ388928 and FJ388929) from Cyprus whose nearly complete coding region sequences were recently released were very close to CRF45_AKU. 24 In gag and env they seem identical to CRF45_AKU, whereas they are like subtype A in the pol region, lacking the K and undetermined subtype segments from CRF45_AKU.
The main objective of this study was to determine whether some HIV-1 strains from Equatorial Africa that clustered with HIV-1_MAL and HIV-1_NOGIL in gag and pol and with the recently described 04FR.AUK 8 in env represent a novel CRF, and to define their recombinant structure. We also examined the extent to which these viruses circulate in the current HIV-1 group M epidemic around the world and we reanalyzed the mosaic structure of HIV-1_MAL and HIV-1_NOGIL3 9,10 isolated in 1985 and in the early 1990s, respectively. Four such strains, for which sufficient material was available, were identified and full-length sequenced.
Phylogenetic and recombination analyses showed that all five strains had the same recombinant structure and thus represent a new CRF, CRF45_AKU, composed of alternating fragments of subtypes A, K, and undefined segments. More than 60% of the genomic structure is composed of subtype A, although it is different from the A1 to A5 subsubtypes as well as from A sequences present in known CRFs or other A variant strains. 7,14,15 However, it is now well known that the subtype A radiation is composed of very divergent lineages and is characterized by the highest intrasubtype genetic variability. 15 Subtype K represents approximately 25% of the mosaic pattern and the remaining 15% were undefined subtype/CRF “U” segments. However, two of these U segments were monophyletically related to U fragments from CRF04_cpx in pol and from CRF09_cpx strains in env. Moreover, CRF09_cpx strains also clustered with the CRF45_AKU strains in two additional regions: gag and the major part of the protease genes in which the strains were subtype A, and a small region of the RT in which they were subtype K.
Globally, the three segments in which the CRF45_AKU and CRF09_cpx strains seemed monophyletic represent 2200 nucleotides, suggesting a partial common evolutionary history for CRF45_AKU and CRF09_cpx. CRF09_cpx strains, described in West Africa and Cameroon; they are known to be very complex and until now the final recombinant structure of these strains is still not entirely resolved (
The recombination and phylogenetic analyses from our study demonstrated that the CRF45_AKU lineage could represent the common ancestor of HIV-1_MAL and HIV-1_NOGIL3. The NOGIL3 strain could result from a recombination between CRF45_AKU and subtype H; the HIV-1_MAL strain has a more complex structure and is the result of recombination between the CRF45_AKU and D subtype. Taking into account the date at which HIV-1_MAL was isolated, the parental CRF45_AKU lineage probably existed before 1981. 9,10 Indeed, the high intrasubtype genetic distances also suggest that the CRF45_AKU strains circulated for a long time.
Finally, we screened existing databases in order to find sequences clustering with or sharing the same structure as CRF45_AKU and found numerous partial sequences from HIV-1 strains in Africa, Europe, as well as China and Argentina that clustered within the CRF45_AKU lineage. In Africa, the highest frequency was seen in Equatorial Africa (Gabon, Republic of Congo, DRC, Cameroon, and CAR), but they were also present in Western Africa and in Europe, mainly Southern Europe. Although the majority of the sequences that matched were derived from the RT region, and eventually also from other genomic regions such as gag and env, more sequences are needed to allow a conclusion to be drawn about the real prevalence of CRF45_AKU strains. However, these results allowed us to conclude that fragments of this lineage are currently circulating worldwide.
This study also illustrated the high viral diversity of HIV-1 strains originating from Central Africa and with the continuing spread of HIV-1, the possibility that this diversity may be exported elsewhere. Factors that influence the spread of particular subtypes or CRFs in different geographic regions are not really understood. In the HIV-1 epidemic and evolution, some viruses become founder strains for local or global epidemics, while others are not maintained in the population. It is therefore important to be able to track the origin and spread of HIV-1 variants in the context of vaccine development, surveillance of antiretroviral drug resistance, and treatment scale-up.
Sequence Data
The sequences have been deposited in the EMBL database under accession numbers FN392874 to FN392877.
Footnotes
Disclosure Statement
No competing financial interests exist.