
Abstract
Analysis of 3555 HIV-seropositive specimens, collected in Cameroon from 2002 to 2006, led to the identification of four HIV-1 group N infections based on differential seroreactivity to HIV env-derived peptides and proteins and confirmation by nucleic acid amplification. Group N prevalence continues to be low accounting for only 0.1% of HIV infections in Cameroon. Near full-length genomic sequences were obtained from viral RNA or proviral DNA by PCR amplification of overlapping fragments for three isolates, 06CM-U14296, 06CM-U14842, and 02CM-SJGddd. Two genome segments, partial pol and env–nef, were obtained from viral RNA for the fourth isolate, 02CM-TIM0217. With the four group N isolates identified in this study and group N sequences previously reported, eight near full-length and five partial genome sequences are now available. Despite genetic divergence from HIV-1 group M and O, all of the group N infections evaluated by five commercial HIV immunoassays were detected.
H
Simian immunodeficiency viruses (SIV) have been transmitted into humans at least three times to give rise to HIV-1 groups M, N, and O and at least eight times to give rise to HIV-2 groups A–H. 7 Cameroon is the natural habitat for the nonhuman primates that harbor the SIV strains most closely related to the HIV-1 groups. Chimpanzees, Pan troglodytes troglodytes, residing in the south central region of Cameroon harbor an SIV most closely related to HIV-1 group N while P. t. troglodytes in the southeast corner of Cameroon harbor an SIV strain more closely related to group M. 8 Western gorillas, Gorilla gorilla gorilla, in communities located in south central and the southwest corner of Cameroon harbor an SIV strain (SIVgor) distinct from but related to HIV-1 group O. 9,10 In addition to being a country endemic for HIV-1 groups M, N, and O, Cameroon has a high level of HIV strain diversity making it a likely place to identify new and/or rare HIV variants. Very recently, a new HIV-1 strain designated group P that appears to be derived from SIVgor was identified in a Cameroonian woman. 11
To better understand the prevalence of group N, plasma specimens were collected from HIV-infected Cameroonians and screened to identify group N isolates. We report the identification of four HIV-1 group N infections using a serological algorithm developed to discriminate between HIV-1 groups M, N, and O, as well as to identify new HIV strains. 3 Characterization of the group N-infected specimens included obtaining near full-length genome sequences for three of the group N isolates and partial genome sequences for the fourth isolate. In addition, specimens from the group N-infected individuals were tested with five commercially available HIV immunoassays to evaluate the ability of the assay to detect group N infections.
During 2005 and 2006, blood specimens were collected at Jamot Hospital, Faculty of Medicine and Sciences Biomedical, and Centre Hospitalier et Universitaire in Yaoundé, Cameroon from hospital and tuberculosis clinic patients. Blood specimens were also obtained from individuals attending hospitals and clinics in the Northwest, Southwest, and Western Provinces of Cameroon from 2002 to 2005. Specimens were initially screened for HIV infection in Cameroon and at Abbott Diagnostics using at least two commercially available tests: Murex HIV Ag/Ab Combination EIA (List GE41/42; Abbott Laboratories, Dartford, UK), Determine HIV-1/2 (List 7D23; Abbott Laboratories, Tokyo, Japan), and HIVAB HIV-1/HIV-2 (rDNA) EIA (List 3A77; Abbott Diagnostics, Abbott Park, IL). Any specimen reactive in at least one of the tests was further analyzed using a research MO2N rapid test 12 and peptide enzyme-linked immunoassays (PEIA). The PEIAs use peptides derived from the immunodominant region (IDR) of env gp41 and the V3 loop region of env gp120 of HIV-1 groups M, N, and O and SIVcpz strains GAB and ANT and have been described previously. 13
Of 3555 HIV-seropositive specimens from Yaoundé and the three provinces, the serological screening algorithm identified four specimens as HIV-1 group N. All four specimens, SJG-2002-ddd, TIM-2002-B-0217, U14296, and U14842, were reactive to the HIV-1 group N and SIVcpz GAB peptides in the IDR and V3 PEIAs and the three that were tested in the MO2N rapid test were reactive to the group N recombinant protein (Table 1). The low prevalence of group N in Cameroon, 0.1% (4 of 3555), is consistent with previous reports. 1 –3 Specimens U14296 and U14842 were collected in 2006 from patients attending the Jamot Hospital Tuberculosis Clinic in Yaoundé. U14296 was collected from a 47-year-old female and U14842 was collected from a 48-year-old male. SJG-2002-ddd was collected at St. John of God Hospital in Nguti, Southwest Province in 2002. TIM-2002-B-0217 was collected in the Southwest Province in 2002. No patient data are available for SJG-2002-ddd and TIM-2002-B-0217.
Test used in serological screening algorithm to identify group N infections. s/co, signal to cutoff ratio, values >1.0 are reactive.
Specimen diluted 1:10 before testing.
Specimen diluted 1:200 before testing except specimen1131–03 that was diluted 1:50.
Reactivity to peptides listed in order of decreasing signal. M, group M; N, group N; Gab, SIVcpz GAB; Ant, SIVcpz ANT.
na, data not available.
Group N specimen reported previously.3,5,6
To confirm the presence of group N virus, nucleic acids extracted from either plasma or whole blood were used to amplify viral sequences. Conditions for nucleic acid extraction, amplification, sequencing, and phylogenetic analysis have been described previously. 3,5 Briefly, reverse transcriptase polymerase chain reaction (RT-PCR) amplification of the IDR region using primers that amplify HIV-1 groups M and N was performed using the QIAGEN One Step RT-PCR Kit (Qiagen, Valencia, CA) followed by direct sequencing of the IDR fragments. Phylogenetic analysis of the IDR sequences confirmed all four patients were infected with HIV-1 group N (data not shown).
Near full-length genomes were obtained by PCR amplification of overlapping fragments using proviral DNA extracted from whole blood of patients U14296 and U14842 and viral RNA extracted from plasma of patient SJG-2002-ddd. DNA sequences were named using the following convention: year collected and two letter country code followed by a dash and specimen ID, i.e., the genome sequence amplified from patient U14296 is designated 06CM-U14296. The assembled genomes of 06CM-U14296, 06CM-U14842, and 02CM-SJGddd are 8545 nucleotides (nts), 8637 nts, and 8615 nts, respectively, and encode open reading frames for all of the HIV structural and regulatory genes. Using viral RNA extracted from plasma of TIM-2002-B-0217, only two genome fragments were amplified, env–nef (1322 nts) and pol integrase (828 nts). An examination of the pol protease-RT and integrase sequences of the group N viruses found no polymorphisms known to confer resistance to antiretroviral therapy in HIV-1 group M strains (data not shown). 14
A phylogenetic tree constructed from an alignment of 06CM-U14296, 06CM-U14842, and 02CM-SJGddd genomes and HIV-1 group M, N, and O reference sequences 15 confirmed the three isolates as group N (Fig. 1). The three isolates fall within the group N cluster with all unlinked sequences being approximately equidistant from each other (isolates 04CM-1015-04 and 04CM-1131-03 obtained from a husband and wife are linked 5 ). Simplot analysis (SimPlot v3.5.1; S. Ray, Johns Hopkins University, Baltimore, MD) of the three new group N sequences and five genome sequences previously reported 1,3-6 shows the eight group N strains are highly similar to each other across the genome and the sequences do not contain any segments of group M, group O, or SIVcpz (data not shown). The high level of sequence similarity and low interisolate genetic distances for the group N isolates are consistent with group N being more recently transmitted into humans than HIV-1 groups M and O.
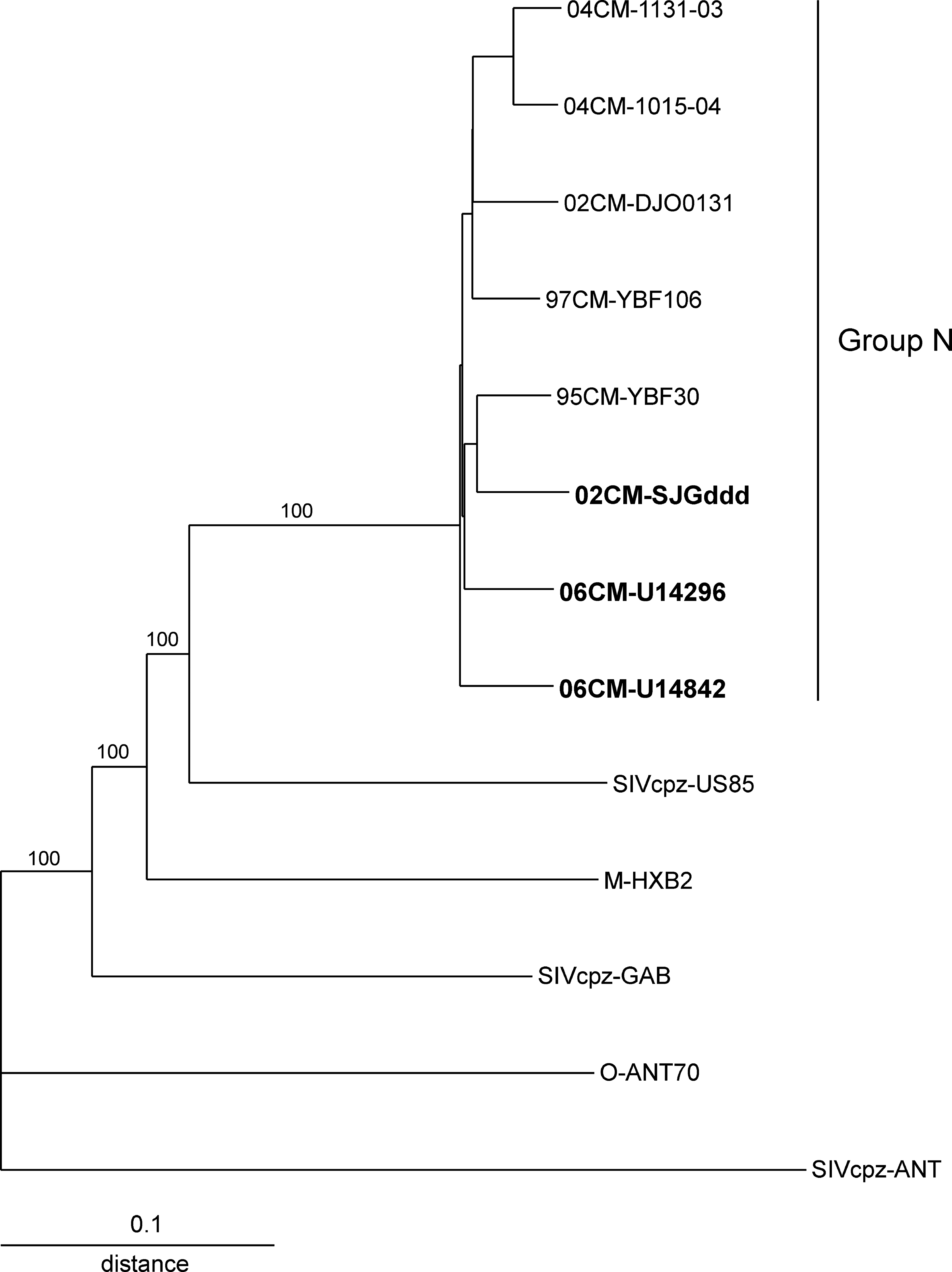
Phylogenetic tree derived from the alignment of 06CM-U14296, 06CM-U14842, and 02CM-SJGddd genome sequences and HIV-1 group M, N, and O, and SIVcpz reference sequences. Sequences were aligned using Megalign (Lasergene, v. 7.2.1, DNASTAR, Inc., Madison, WI) and manually edited. Phylogenetic analysis was performed with PHYLIP software (v. 3.573c for windows, J. Felsenstein, University of Washington, Seattle, WA) using Dnadist (Kimura two-parameter) to estimate genetic distances, Neighbor (neighbor-joining method) for phylogenetic relationships, and Seqboot for branch reproducibility. Alignment was 8033 nts in length after gaps were stripped. SIVcpzANT was used as the outgroup. Bootstrap values are shown for selected branches.
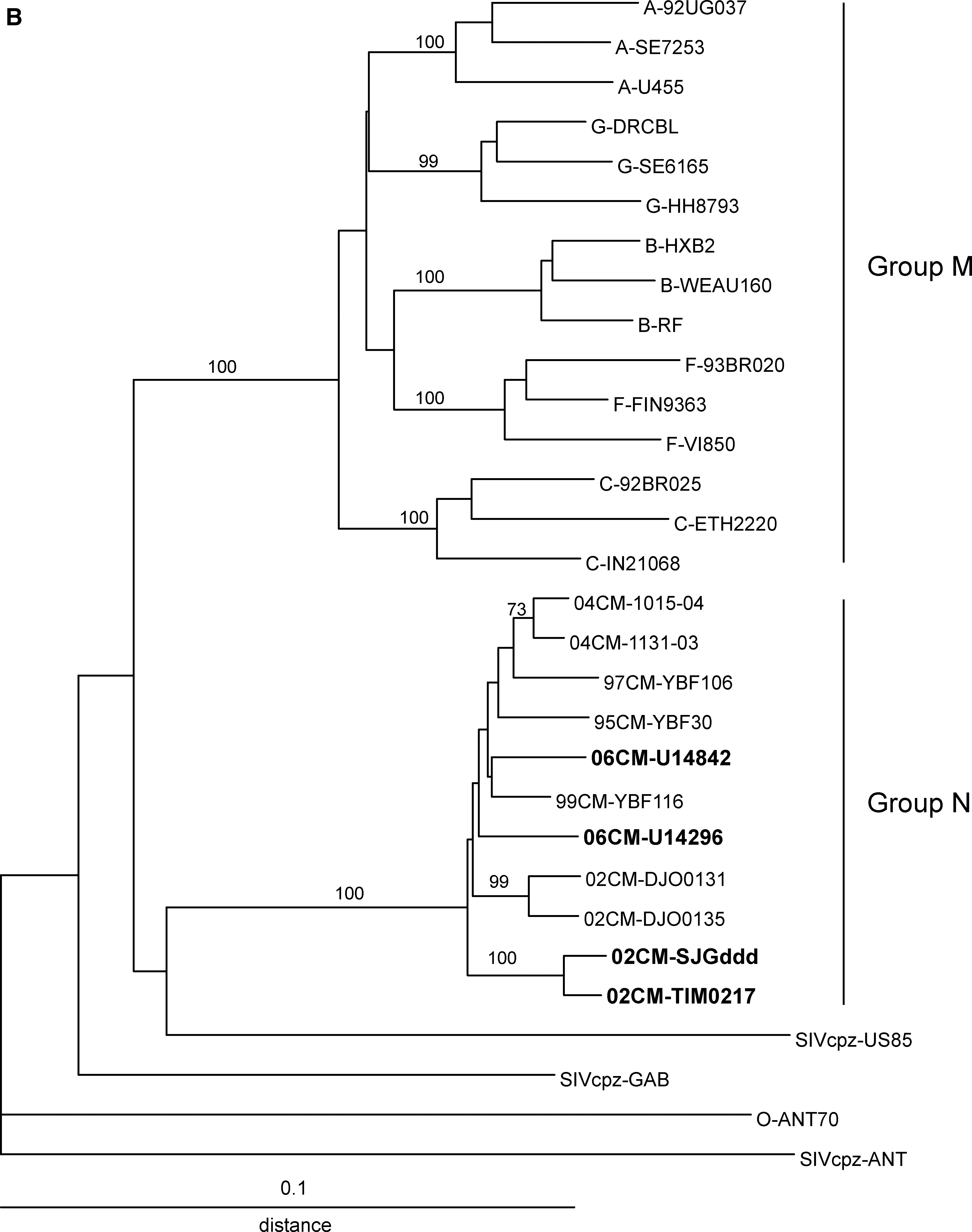
Phylogenetic trees were constructed from alignments of partial genome sequences encoding env gp41 IDR and pol integrase to allow inclusion of the four new isolates and additional group N isolates reported previously 1,3 –6 . The IDR tree (Fig. 2A) includes sequences from 12 group N isolates and the pol tree (Fig. 2B) includes 11 group N sequences. These trees illustrate the low genetic diversity between the group N isolates; genetic distances between unlinked group N isolates ranged from 0.024 to 0.091 and 0.021 to 0.052 for IDR and integrase, respectively. The intragroup distances for group N are less than or equal to the intrasubtype distances for group M. In both trees, the two group N isolates known to be linked, 04CM-1015-04 and 04CM-1131-03, 5 and two isolates that are suspected of being linked, 02CM-DJO0131 and 02CM-DJO0135, 6 form pairs and are separated by short genetic distances: 0.016 and 0.010 for IDR and 0.011 and 0.018 for integrase, respectively. Similarly, 02CM-SJGddd and 02CM-TIM0217 form a pair separated by short branch lengths suggesting these infections are linked: an interisolate genetic distance of 0.0026 for IDR and 0.014 for integrase. However, in the absence of patient information linkage cannot be confirmed.

Phylogenetic trees derived from sequence alignments of env gp41 IDR (
An alignment of the IDR amino acid sequences from 12 group N isolates shows a high level of sequence conservation; in the 35 amino acid cluster I epitope in the IDR, only six positions have amino acid heterogeneity (Fig. 3A). One sequence, 06CM-U14296, has an atypical proline residue in the Cys–Cys loop. Despite this substitution, antibodies to this virus bound group N, group M, and SIVcpzGAB IDR peptides that have the consensus threonine at this position (Table 1). A comparison of the consensus sequences for groups N and M shows conservation at 24 of the 35 amino acid positions whereas groups N and O consensus sequences are conserved at 19 positions. A total of 16 positions are conserved across the consensus sequences for all groups. Similarly, the amino acid sequence of the V3 loop is relatively conserved across eight group N isolates with only 6 of 35 positions showing sequence heterogeneity (Fig. 3B). A comparison of the groups N and M consensus sequences shows 22 conserved residues and groups N and O have 12 conserved residues; at 11 positions, all three HIV-1 groups are conserved. The consensus sequence for the tip of the V3 loop is GPAM, GPQA, and GPMA for groups N, M, and O, respectively. In the V3 loop there is one glycosylation site near the N-terminus that is conserved in all groups.

Amino acid consensus sequences for HIV-1 group M, N, and O. (
With the four infections in this study and four reported previously, 3,5,6 we have identified and confirmed a total of eight HIV-1 group N infections in Cameroon from specimens collected from 1999 through 2006. Since data on the seroreactivity of HIV-1 group N infections are limited, plasma specimens from the eight infected individuals were tested using five commercially available HIV immunoassays that detect antibody to HIV-1 group M and HIV-2; four also detect antibody to group O. Initial screening of neat specimens was performed using Murex HIV Ag/Ab Combination EIA, Determine HIV-1/2, and HIVAB HIV-1/HIV-2 (rDNA) EIA, and all tested specimens were reactive in these assays (Table 1). Because specimen volume was limited, additional testing was performed using specimens diluted in negative human plasma. At dilutions of 1:10 up to 1:200, all seven specimens tested were reactive in the ARCHITECT HIV Ag/Ab Combo (List 4J27; Abbott Diagnostics, Wiesbaden, Germany), Murex HIV Ag/Ab Combination, and Murex HIV Ab EIA (List GE94/95) assays (Table 1).
The group N infected specimens were all reactive in commercially available HIV immunoassays that detect HIV-1 group M and group O and HIV-2. However, it should be noted that only specimens seropositive for HIV were screened for group N. Thus, any group N infections not identified as HIV seropositive by at least one of the three initial serological assays were not included in the screening algorithm. Despite this caveat, the relative conservation of the immunodominant region of env gp41 between group M and group N may account for the detection of antibodies to group N. Six of seven group N-infected specimens detected by the MO2N test showed cross-reactivity to the HIV-1 group M recombinant env protein and four of eight cross reacted to the group M peptide in the IDR PEIA (Table 1). In contrast, none of the group N infections showed cross-reactivity to group M in the V3 PEIA or to group O env protein and peptides.
The serological screening algorithm (identifying HIV-seropositive specimens followed by screening with PEIAs) is an effective tool for identifying HIV-1 group N infections. The eight HIV-1 group N specimens exhibited similar patterns of reactivity in the PEIAs characterized by preferential binding to the group N and SIVcpz GAB peptides and no cross-reactivity to the group O peptides (Table 1).
In conclusion, the prevalence of HIV-1 group N in Cameroon continues to be low. However, group N is a pathogenic virus that can cause AIDS. 1,2,5 Continued surveillance studies are warranted to monitor changes in the prevalence of HIV-1 group N, to detect potential spread outside of Cameroon and to identify the emergence of additional HIV groups.
Footnotes
Acknowledgments
GenBank accession numbers for the sequences reported here are GQ324958–GQ324962. We thank P. Swanson, B. Harris, V. Holzmayer, and J Yamaguchi for reviewing the manuscript.
Disclosure Statement
No competing financial interests exist.