
Abstract
Although subtype B strains are still predominant in France, non-B viruses have been isolated from 26% of patients with a primary HIV-1 infection in 2005–2006. The objective of this study was to characterize recombinant-HIV-1 strains by a subtyping based on the phylogenetic analysis of both pol and env sequences. We studied 591 patients who were part of the French PRIMO-Cohort between 1997 and 2007. The RT and V3 regions were sequenced and phylogenetic analyses were performed. Phylogenetic analyses showed concordant subtype results for 91.7% of viruses: 71.6% of the viruses were subtype B and 28.4% belonged to non-B subtypes or circulating recombinant forms (CRFs). Forty-nine strains showed different phylogenies between the two genes (pol and env), indicating that recombinations were observed in 8.3% of the cases. These recombinants were observed in patients from sub-Saharan Africa (28.3%) and in white patients (6.3%). Moreover, among the 49 recombinant viruses, 15 (30.6%) contained a B sequence in the pol or in the env gene compared to 34 (69.4%), which contained non-B or CRF sequences. Twenty-six different recombination patterns involving subtypes, CRFs, or undetermined strains were observed. We have reported the occurrence of new recombinant forms between the two major viral types of strains circulating in France: subtype B and CRF02_AG. Our study confirms a high HIV-1 diversity among patients infected in France within the past 10 years, with a high proportion of non-B strains and the circulation of complex recombinant strains among both sub-Saharan patients and French patients.
H
The objective of this report was to study the genetic diversity and molecular epidemiology of HIV-1 in France by identifying the proportion of recombinant strains with different genetic subtypes, based on the pol (RT) and env sequence analyses, in 591 patients infected in 1997–2007 and enrolled in the French ANRS PRIMO Cohort.
The study population included patients with PHI who were enrolled in the multicenter ANRS CO06 PRIMO Cohort. 8 Patients were enrolled if they had been infected with HIV less than 6 months ago. At the enrollment stage, a physical examination was performed on the patients and blood samples [plasma and peripheral blood mononuclear cells (PBMCs)] were collected. As genotypic resistance tests were performed each time a patient was included in the cohort, RT sequences were collected.
Briefly, plasma HIV-1 RNA was extracted and amplified using the consensus technique of the AC11 ANRS Resistance group (
Phylogenetic trees were built using the neighbor-joining method with 1000 bootstrapped data sets (PHYLIP package). 13 Bootstrap values ≥700 were considered as significant for belonging to a subtype or a CRF. TreeView Win16 was used to construct the trees. 14 To analyze the recombinant structure of viruses undetermined after phylogenetic analysis, several additional analyses were performed. Simplot 3.5.1 software was used to determine the percentage of similarity between selected pairs of sequences and to calculate bootscan plots, by performing bootscanning on parsimony trees using SEQBOOT, DNADIST (with Kimura's two-parameter method and F84 model of maximum likelihood method, transition/transversion ratio = 2.0), NEIGHBOR, and CONSENSE from the Phylip Package. 15 In the similarity and bootscan plots, the new sequences were compared with consensus sequences (50% threshold) of the no-recombinant subtypes and some CRF reference strains. Statistical comparisons were based on the Student's t test or Wilcoxon nonparametric test for continuous variables. Percentages were compared by using the chi-square test or Fisher's exact test. Data were analyzed using SAS software (SAS Institute Inc.).
Between 1996 and 2007, 870 patients were enrolled in the French ANRS PRIMO Cohort. In this substudy, we included 591 patients with available sequences centralized between 1997 and 2007. Of the 591 strains, 542 (91.7%) provided a concordant phylogenetic analysis for the env and pol genes whereas 49 strains showed different phylogenies between the two genes. This indicates that recombinations have occurred, which has led to the circulation of complex recombinants in 8.3% of the cases. As the pol and env sequences were obtained from plasma RNA and provirus DNA from PBMCs, respectively, we cannot ensure that the amplified sequences of the two different genomic regions are derivative of the same viral clone. Thus, we chose to label these recombinants as “putative recombinants.”
Among the 542 concordant strains, 388 belonged to subtype B (71.6%) and 154 (28.4%) to non-B subtypes. We observed a high diversity among the 154 non-B viruses; 91 (59.1%) belonged to CRF02_AG, 17 (11%) to subtype A, 10 (6.5%) to C, 8 (5.2%) to G, 7 (4.5%) to D, 7 (4.5%) to CRF42_BF, 4 (2.6%) to F, 3 (1.9%) to CRF01_AE, 2 (1.3%) to CRF27_cpx, 16 1 to CRF06_cpx (0.6%), and 1 to CRF09_cpx (0.6%). Three (1.9%) viruses remained undetermined in both genes as they did not cluster with any known subtype or CRF. We observed 49 complex recombinant viruses, 31 (SGRs) and 18 viruses involving recombination between two subtypes, overall with new different recombination patterns (Table 1).
Each patient was identified with the year of inclusion, FR for France, and the letter corresponding to each patient. MSM; men having sex with men: NRTI; nucleoside reverse transcriptase inhibitor.
The phylogenetic analysis of the 49 pol sequences (Fig. 1) revealed that 10 strains (10/49, 20.4%) clustered with subtype B sequences, 13 (26.5%) with CRF02_AG, 5 (10.2%) with CRF06_cpx, 2 (4.1%) with CRF11_cpx, 2 (4.1%) with CRF12_BF, 2 (4.1%) with subtype A, 1 (2%) with F, 1 (2%) with CRF09_cpx, and 1 (2%) with CRF18_cpx. Twelve (24.5%) viruses remained undetermined after analysis of the pol sequences, as they did not cluster with any known subtype/CRF with a significant bootstrap value (≥700). The phylogenetic analysis of the 49 env sequences indicated that 16 strains (32.7%) clustered with subtype A, 7 (14.3%) with CRF02_AG, 5 (10.2%) with subtype G, 5 (10.2%) with subtype B, 5 (10.2%) with C, 3 (6.1%) with D, 3 (6.1%) with F, 2 (4.1%) with CRF06_cpx, and 1 (2%) with CRF33/34_01B. Only two strains (4.1%) remained undetermined following the phylogenetic analysis of the env region. The baseline characteristics of the 49 patients with complex recombinant viruses are summarized in Table 1.
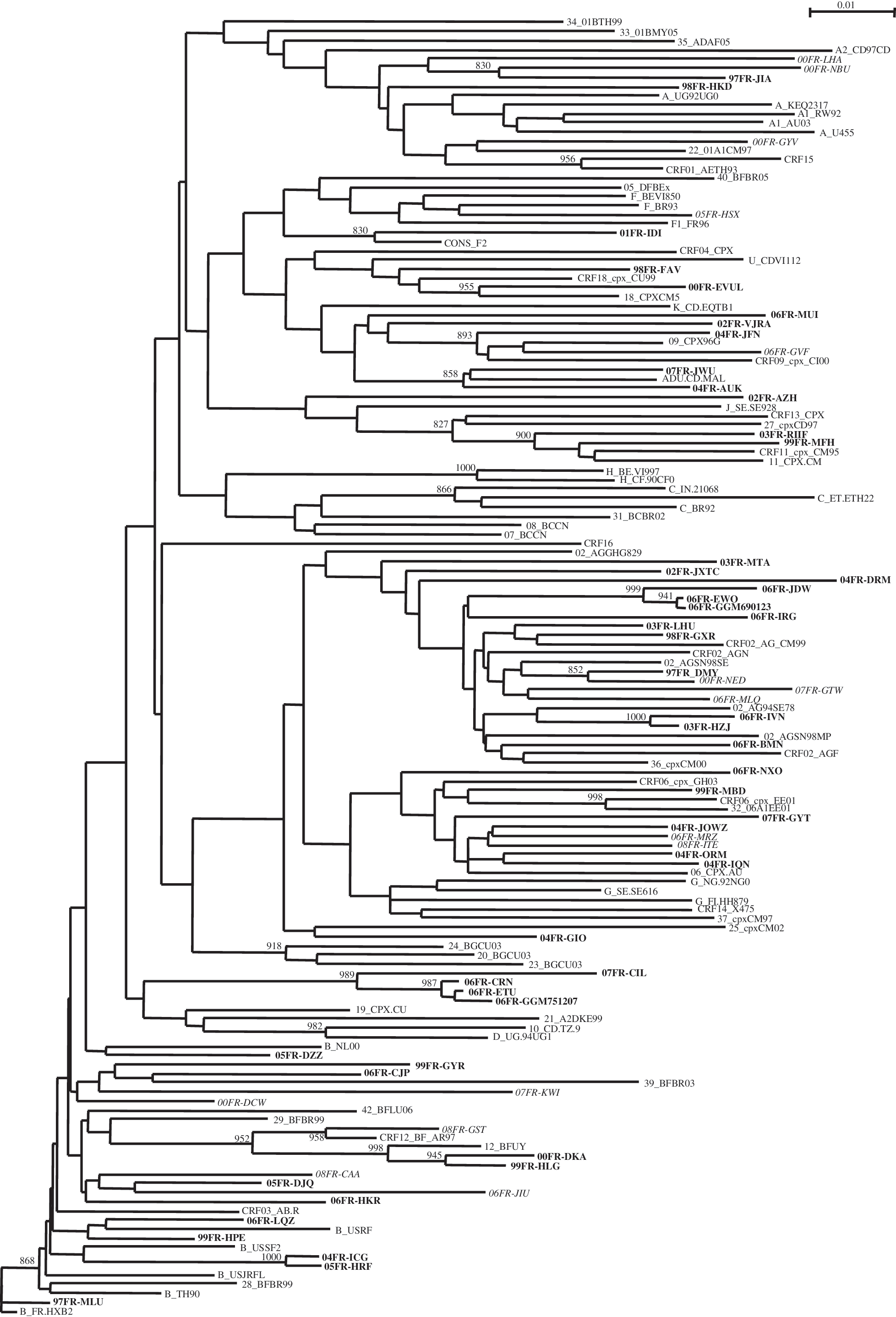
Phylogenetic relationships of RT sequences based on sequence comparisons of the 49 complex recombinants viruses (in bold) with 66 previously reported sequences representatives of group M including reference sequences of subtypes, subsubtypes, and at least one of each CRF sequence available in the HIV database (until CRF 42) (
Cases of primary infection occurred with recombinants from the beginning of the PRIMO Cohort (1997) to 2007. The median CD4 cell count and the median HIV-1 RNA amounted to 470 cells/mm3 (range 128–938) and 4.90 log10 copies/ml (range 1.79–8.33), respectively. Among the 49 recombinant viruses, 26 different patterns of recombination were identified; they involved subtypes, CRFs, and undetermined strains. We described 31 SGRs and 18 viruses involving a recombination between two subtypes. Subtype A and/or CRF 02_AG were involved in most of the recombinant events (29/49, 59%). Among the 49 patients infected with a complex recombinant virus, 30 (61%) were from France: 14 male patients had been infected through homosexual intercourse (“men having sex with men,” MSM), 15 patients through heterosexual contact (9 women and 6 men), and 1 woman through intravenous drug use (IVDU) (Table 1). A significantly higher frequency of complex recombinant viruses was observed in patients from sub-Saharan Africa (15/53, 28.3%) compared to white patients (34/538, 6.3%, p < 0.0001).
The baseline characteristics of the 154 patients infected with a concordant non-B strain were compared to those of the 49 patients infected with a complex recombinant virus (Table 2). Following adjustments related to sex, patient origin, and time from infection, no significant difference regarding the CD4 cell count, HIV-1 RNA, and HIV-1 DNA was observed between the two groups of patients.
IVDU, intravenous drug user.
Following the phylogenetic analysis of the pol gene, 398 strains were identified as B subtypes. Among these 398 strains, 388 (97.5%) had a concordant subtype in the env gene and 10 (2.5%) of them belonged to a non-B subtype. Whereas among the 193 non-B strains, 154 were identified as non-B concordant subtypes or CRFs and 39 strains as complex recombinant viruses (20.2%). Moreover, among the 49 viruses, 15 (30.6%) contained a B sequence in the pol or env gene compared to 34 (69.4%), which contained non-B or CRF sequences. All the 15 patients except 1 with a virus containing a B sequence were white. Among the 20 women infected with a complex virus, only 4 (20%) were infected with a virus containing a B sequence whereas 10 were found among the 17 MSM (59%), p < 0.005. In this case, MSM were significantly more often infected with a virus containing a B sequence than women (odds ratio: 2.95).
The bootscan analysis performed on the 12 strains remained undetermined after the phylogenetic analysis of the pol gene revealed several patterns of recombination (Table 1): 98FR-FAV: F2/K, 02FR-AZH: F2/J, 02FR-VJRA: A/G/F2, 04FR-GIO: G/D, 04FR-AUK: A/K, 06FR-NXO: CRF06/G, 06FR-MUI: A/G/F, and 07FR-JWU: A/K. Four strains (06FR-CRN, 06FR-ETU, 06FR-GGM, and 07FR-CIL), which remained undetermined after the pol analysis, were classified as C subtypes in the env sequence analysis. The phylogenetic tree revealed that these four viruses clustered together with a bootstrap of 989. These four viruses were isolated in four MSM infected in the Parisian area and we cannot exclude a possible epidemiological link between the patients. The bootscan analysis of these four viruses revealed a B/C pattern of recombination. The full-length sequence of one of the viruses (06FR-CRN) revealed a B/C/U recombinant structure involving a Brazilian C strain. 17 Finally, two strains (04FR-AUK and 07FR-JWU) clustered in the pol analysis with MAL, one of the earliest African HIV-1 strains, previously identified as an A/D/K/U recombinant virus. 11 The 04FR-AUK and 07FR-JWU env sequences belonged to subtype A whereas the MAL envelope sequence clustered in subtype D.
In this article we reported a high genetic diversity of HIV-1 strains among 591 patients diagnosed at the time of PHI in France between 1997 and 2007, which is one of the largest series to date. More than 90% of the viruses provided a concordant phylogenetic analysis between the env and pol genes: 71.6% belonged to subtype B and 28.4% to non-B subtypes with a great viral diversity: 12 different subtypes or CRFs were observed. The frequency of the CRF02_AG strains remained the highest among non-B viruses. This is most probably due to the successive migratory flows from French-speaking West African countries. Complex recombinants were identified in 8.3% of the cases as 49 strains showed different phylogenies between the two genes (pol and env), thus implying that recombinations have occurred.
As it is known that non-B HIV-1 increases over time in France and in Europe, our study provides new insights on the HIV epidemiology in France with a more detailed description of the circulating variants. The frequency of 28.3% of complex recombinants observed in patients from sub-Saharan Africa was similar to the prevalence of 23–47.1% new recombinant forms reported in Nigeria and Angola. 18,19 Moreover, our results for African patients comply with the recent study performed by Yebra et al., which reported 37% of intersubtype recombinants among 66 African patients diagnosed in Spain during 2005–2007. 20 Similarly, following the phylogenetic analysis of the pol gene, 20.2% of complex recombinants were discovered in patients infected with a non-B strain. This result is similar to the results obtained for 84 French patients chronically infected with non-B viruses for which 21.4% of intersubtype recombinant strains were observed. 21 However, our 20.2% rate is higher than the rate obtained in the recent retrospective Spanish study, which reported 14 new recombinant strains in 171 infected subjects with non-B strains (8.2%). 22
Despite a significantly higher frequency of complex recombinant strains in patients from sub-Saharan Africa, originated from countries where all the subtypes and most of the CRFs cocirculate, we also observed a high diversity in French white patients [30/49 (61%) of patients infected with a complex recombinant]. This result shows that the increasing viral diversity is currently being observed in France, not only in sub-Saharan patients but also among white patients diagnosed at the time of PHI.
Our results illustrate the circulation of old HIV-1 strains in France—the recent isolation of two strains (04FR-AUK and 07FR-JWU), which significantly clustered on the pol sequence with the ancestral strain MAL. The diversity of HIV-1 in France is not due only to successive migratory flows from the French-speaking African countries; our study has described several strains introduced by Asian (pure or recombinant CRF01_AE and CRF33/34 strains) and South American countries (CRF42_BF, B/F, and CRF12/F recombinant strains, the cluster of four URF B/C/U derived from a Brazilian C virus). The emergence, since 2005, of new recombinant forms among the two major types of strains circulating in France—subtype B and CRF02_AG—must be noted.
It is interesting to note that 14 of the 15 viruses containing a B sequence have infected white patients. It is impossible to determine if these recombinant viruses were introduced in France from outside or if the variants were the result of local events of recombination. We could at least suggest that considering the fact that among the 15 viruses containing a B sequence, 14 patients were white and were infected in France, the recombination events could occur in France where subtype B is predominant.
Interestingly, in this study, we have reported the variation of the diversity associating not only new strains (CRF02/B) but also old recombinant strains (“MAL like” 04FR-AUK and 07FR-JWU). Using another phylogeographic approach, Paraskevis et al. recently described a study tracing HIV-1 subtype B mobility in Europe. 23 They have reported that in most European countries, the HIV epidemic was introduced by multiple sources and subsequently spread within local networks.
Despite the diversity of the pol gene, this viral region has been successfully used for HIV-1 subtyping. 9 With few exceptions, the CRFs described so far have shown breakpoints in the pol gene, which therefore can be considered as a possible “hot spot” for recombination. However, our study has shown that associating the subtyping on two genes improves the frequency assessment of recombinant strains.
To conclude, our study confirms the high HIV-1 diversity in patients diagnosed at the time of PHI, with a circulation of complex recombinant strains including second generation recombinants in France among sub-Saharan patients as well as among French patients. Accurate classification of new recombinants is important to determine whether these strains may have a possible founder effect in France. Our results reinforce French guidelines that recommend performing genotypic resistance tests at the time of PHI to survey the frequency of resistant strains as well as the molecular epidemiological HIV-1 diversity.
Nucleotide Accession Numbers
GenBank accession numbers for the sequences reported in the study are GQ410023 to GQ410071 for the reverse transcriptase gene and GQ409974 to GQ410022 for the env gene.
Footnotes
Acknowledgments
The authors are indebted to the patients enrolled in the cohort. Without their participation, none of these studies would have been possible. The full list of investigators for the French Primo Study group can be found at
Author Disclosure Statement
No competing financial interests exist.