
Abstract
The recombinant vaccine, tgAAC09, based on an adeno-associated virus serotype 2 (AAV2) vector encoding HIV-1 subtype C Gag, protease, and part of reverse transcriptase, induced robust T cell and antibody responses in nonhuman primates. In a previous phase I study in 80 healthy HIV-seronegative European and Indian adults, the vaccine was generally safe, well tolerated, and modestly immunogenic when administered once at doses up to 3 × 1011 DRP. This phase II double-blind, randomized, placebo-controlled trial tested two administrations and a higher dosage of tgAAC009. Ninety-one healthy HIV-seronegative adults from three African countries were given one of three dosage levels of tgAAC09 (3 × 1010, 3 × 1011, or 3 × 1012 DRP) intramuscularly, either at a 6- or 12-month interval; follow-up was 18 months. Overall, 65% and 57% of vaccine recipients experienced local and systemic signs and symptoms, respectively, most being mild. Frequency and severity were not dose related and were similar to those in placebo recipients. No vaccine-related serious adverse events were reported. Overall, HIV-specific T cell responses were detected by IFN-γ ELISPOT in 17/69 (25%) vaccine recipients with 38% (10/26) responders in the highest dosage group. The response rate improved significantly with boosting at 6, but not 12 months, in the 3 × 1011 and 3 × 1012 dosage groups only. Neutralizing antibody titers to the AAV2 did not alter the frequency of immune responses to HIV. Two doses of tgAAC09 were well tolerated at the dosage levels given. Fewer than half the recipients of the highest vaccine dosage, 3 × 1012 DRP, had T cell responses to HIV.
Introduction
T
Recombinant AAV-based (rAAV) vaccines have been shown to be efficacious in nonhuman primates. In a challenge study conducted in macaques using a multicomponent rAAV2 vaccine against simian immunodeficiency virus (SIV), vaccinated macaques had lower peak viral load, lower viral set point, and a lower rate of succumbing to SIV-related illness; by 16 weeks postchallenge, three of eight control animals had died of SIV-related illness, but none of the vaccinated animals died during the 12-month observation period after challenge. 5 Immunogenicity studies of tgAAC09 in macaques demonstrated that a single injection of tgAAC09 induces dose-dependent, specific humoral and cellular immune responses against HIV-1 Gag, which can be boosted by repeat homologous administration 80 weeks after the initial dose (unpublished data).
A phase I double-blind, randomized, placebo-controlled dose-escalating study of tgAAC09 was previously conducted in 80 HIV-seronegative individuals in Europe and India. The vaccine was shown to be generally safe, well tolerated, and modestly immunogenic after one intramuscular injection at dosages up to 3 × 1011 DNase-resistant particles (DRP). 6 The strong T cell and antibody-mediated immune responses observed in macaques were not reproduced in this clinical study, perhaps because the dosages administered in macaques were higher, ranging from 5 to 10 × 1012 DRP for each of the three vaccine components, 5 in contrast to 3 × 1011 DRP for the single component tgAAC09 vaccine given to humans. In the phase I clinical trial there was no evidence of vaccine shedding in urine, semen, and vaginal and nasal secretions collected 1, 3, 14, 56, and 168 days after vaccination with up to 3 × 1011 DRP and the vaccine was not found in peripheral blood mononuclear cells (PBMCs). That study was too small to determine the effect of preexisting immunity to AAV2 on immune responses to the HIV transgene.
The rationale for this phase II study was to investigate whether a higher dosage level than previously tested and a second administration would improve the immunogenicity of the study vaccine. This study evaluated (1) the safety and immunogenicity of two intramuscular injections of tgAAC09 at three dosage levels (3 × 1010 DRP to 3 × 1012 DRP) and two different intervals (6 and 12 months), (2) the impact of preexisting neutralizing antibodies against AAV2 capsid on immunogenicity, and (3) the possibility of vaccine shedding after the first injection of high dosage (3 × 1012 DRP) tgAAC09.
Materials and Methods
Study vaccine
The study vaccine, tgAAC09, consisted of purified particles containing the single-stranded DNA from HIV-1 subtype C genes derived from the South African isolate DU422, encoding Gag-PR-ΔRT proteins, enclosed within a recombinant adeno-associated virus serotype 2 (AAV2) protein capsid. 6 The HIV gag-pro-ΔRT nucleic acid is a synthetic cDNA for HIV-1 gag-protease. This includes the gag and protease coding regions and an additional 306 nucleotides of the HIV genome beyond the protease sequence, followed by a stop codon. This encodes the 101 amino acid N-terminal fragment of the reverse-transcriptase (RT) gene, which does not contain either of the RT active sites. The coding sequences were codon optimized and were expressed from a CMV IE promoter. The expression cassette was flanked by the AAV2 inverted terminal repeat sequences required for packaging. Expression of p24 was confirmed by enzyme-linked immunosorbent assay (ELISA) after in vitro transduction of cells with the vector. The dosage of tgAAC09 is represented in units of DRP.
The vaccine was produced under Current Good Manufacturing Practice (cGMP) guidelines at Targeted Genetics Corporation (Seattle, WA) and was formulated in a sterile isotonic buffered salt solution, which also served as placebo.
Study population
Healthy HIV-seronegative adults were recruited at five clinical trial sites: Uganda [Uganda Virus Research Institute-International AIDS Vaccine Initiative (UVRI-IAVI) HIV Vaccine Program, Entebbe], South Africa [Perinatal HIV Research Unit (PHRU), Soweto, Johannesburg; Desmond Tutu HIV Centre (DTHC), Cape Town; Department of Medical Microbiology, Medunsa, Pretoria], and Zambia [Zambia-Emory HIV Research Program (ZEHRP), Lusaka)]. Eligible participants, between 18 and 50 years of age, provided written informed consent, reported no risk behavior for HIV, and were willing to undergo HIV testing and receive results. Both sexually active men and women agreed to use effective contraceptive methods from screening until at least 4 months after the last vaccination. Health status was determined by medical history, physical examination, and routine laboratory tests.
Study design
This phase II study was a double-blind, randomized, and placebo-controlled study (Table 1). Six cohorts of 13 participants each were to be randomized to receive two intramuscular injections of tgAAC09 at one of three dosages (3 × 1010, 3 × 1011, or 3 × 1012 DRP) or placebo at intervals of either 6 or 12 months in a ratio of 10 vaccine to 3 placebo recipients (Groups A–F). The first six volunteers enrolled at PHRU were to be assigned to the high-dosage/placebo group and consented to provide samples for vaccine shedding studies. Overenrollment of 10% was permitted. Enrollment into these groups was independent of baseline anti-AAV2 neutralizing antibody titers. Through a protocol amendment, a seventh cohort of 13 participants, prescreened for neutralizing antibodies against AAV2 capsid and confirmed to have titers of ≤1/8, was to be randomized to receive two intramuscular injections of either tgAAC09 at a dosage of 3 × 1012 DRP or placebo at a 6-month interval (Group G). Follow-up for safety and immunogenicity was 18 months. The study was approved by all responsible ethical, scientific, and regulatory authorities and conducted according to International Conference on Harmonisation (ICH)—Good Clinical Practice (GCP) and Good Clinical Laboratory Practice (GCLP). 7
Target enrollment was 10:3 in each group. Groups A–F: overenrollment by 8% as permitted by protocol.
Group G participants were prescreened to have anti-AAV2 neutralizing titers ≤1/8. Once the total number of individuals with anti-AAV2 neutralizing titers ≤1/8 enrolled in the protocol (including those chosen for Group G and those incidentally enrolled with low titer in other groups) reached 13, enrollment was halted.
Study procedures
Clinical evaluations
Study participants were monitored throughout the study by interim medical history, physical examination including vital signs, and routine laboratory assessments (complete blood counts, ALT, AST, creatinine, and urinalysis). Solicited local reactogenicity events [pain, tenderness, erythema, edema, skin damage (vesiculation/ulceration), induration, pruritus, formation of crust scab or scar] and systemic signs and symptoms (fever, chills, headache, nausea, vomiting, malaise, fatigue, myalgia, arthralgia, rash, and allergic reaction) were recorded prospectively for 14 (±3) days after each vaccination and graded using the DAIDS Table for Grading the Severity of Adult and Pediatric Adverse Events (Version 6, October 2004). Unsolicited adverse events (AEs), recorded throughout the study, were graded for severity, assessed for relationship to study product, and classified by MedDRA (Medical Dictionary for Regulatory Activities). Solicited local and systemic events that extended more than 14 days after vaccination were recorded as AEs. Severe, very severe, serious, and/or clinically significant laboratory abnormalities were reported as AEs. Safety data were reviewed regularly by an independent Safety Review Board with a representative from each participating country.
HIV testing
At screening, testing for preexisting HIV infection was performed using approved licensed ELISA and rapid tests according to the respective national algorithms. Since tgAAC09 contains HIV gag, a gag-deficient ELISA (Bio-Rad Laboratories Genetic Systems HIV-1/HIV-2 PLUS O EIA) was used during the study to assess for intercurrent HIV infection. In the case of a positive ELISA result, a Western blot and/or a nucleic acid test (Roche Amplicor Version 1.5) were performed to confirm or refute intercurrent HIV infection. At the final study visit, HIV tests were performed using nationally approved HIV test kits to determine the rate of apparent positive HIV test results in participants who had vaccine-induced antibodies, but were not infected with HIV.
Anti-AAV2 capsid neutralizing titers
Serum anti-AAV2 capsid neutralizing titers were measured at baseline, immediately prior to and 2, 4, and 6 months after each vaccination in all groups, and 12 months after the second vaccination in Groups A, C, and E using a microtiter assay. Briefly, serial two-fold dilutions of participant serum samples, a positive control (human serum with known AAV2 neutralizing activity) and a negative control (human serum with no detectable AAV2 neutralizing activity), were incubated in triplicate in a 96-well plate format with wild-type AAV2, and then with human embryonic kidney (HEK) 293 cells infected with adenovirus type 5 as a helper virus. The presence of replicating AAV2 was determined by hybridization, as previously described. 8 The anti-AAV2 capsid neutralizing titer was determined by measuring the inhibition of infectivity as assessed by the absence of hybridizing signal, and was reported as the reciprocal of the lowest dilution at which there were three negative wells. The lowest dilution tested was 1/4. A four-fold or greater rise in anti-AAV2 neutralizing titers postvaccination was considered significant.
Vaccine shedding
Shedding of the vaccine was monitored in the first six participants enrolled into the high-dosage/placebo group at PHRU. Blood, nasal swabs, saliva, urine, and semen or cervicovaginal swabs were collected prior to the first vaccination, and 3 days, 14 days, 2 months, and 6 months after vaccination. The presence of the tgAAC09 vaccine particle was determined by an infectivity assay with a PCR readout. 6 Briefly, the in vitro assay involved incubating the above-mentioned samples with C37 cells containing AAV2 rep and cap sequences in the presence of adenovirus. If tgAAC09 particles were present in the samples, the particles would enter the cells and vaccine DNA in this artificial system would be amplified. The presence of the amplified vaccine DNA was detected by DNA-PCR. The limit of detection of the assay was 3–100 infectious units of tgAAC09 per 10 μl of sample depending on the tissue sample.
Cellular immunogenicity
Vaccine-induced HIV-1-specific T cell responses were assessed immediately prior to and 1, 2, 4, and 6 months after each vaccination in all groups and 12 months after the second vaccination in Groups A, C, and E using a validated ELISPOT assay designed to detect the number of T cells secreting interferon (IFN)-γ, using cryopreserved PBMCs at the IAVI Human Immunology Laboratory (London, UK). Viable PBMCs counted by a Vi-Cell XR counter (Beckman Coulter, UK) were plated at 2 × 105 per well and stimulated in quadruplicate overnight with vaccine-matched peptides at a final concentration of 1.5–2 μg/ml. Peptides (Anaspec, Inc, San Diego, CA) were 15 amino acids (AA) long with an 11 AA overlap. Pools A–D contained 30 peptides each representing segments of Gag. Pool 120 contained 120 peptides representing the entire Gag sequence. Pool 7 contained 22 peptides covering protease. Controls consisted of a 32-peptide pool derived from influenza virus, Epstein–Barr virus, cytomegalovirus (FEC), and phytohemagglutinin (PHA). 9 Responses, expressed as spot forming cells (SFC)/106 PBMCs, were considered positive if all four of the following criteria were fulfilled: (1) mean count greater than 38–60 SFC/106 cells (depending on the peptide pool) above the background, (2) mean count more than four times the mean background SFC count, (3) mean background <55 SFC/106, and (4) coefficient of variation less than 70% across the replicate wells. 10 Immunogenicity data for individuals who acquired intercurrent HIV infection were censored for time points after infection.
Humoral immunogenicity
The vaccine-induced humoral immune response to HIV-1 antigens was evaluated immediately prior to and 2, 4, and 6 months after each vaccination in all groups and 12 months after the second vaccination in groups A, C, and E using a commercially available, indirect diagnostic enzyme immunoassay kit (Bio-Rad Laboratories ELAVIA Ab Ac Ak I) using whole virus antigen.
Statistical considerations
The small sample size was considered to be appropriate for an exploratory study of a novel product while safety and immunogenicity of the vaccine are investigated. To alleviate the effect of heterogeneity and the small sample size in each of the groups (A–G), we planned to combine similar groups for statistical testing of dosage and boost (early vs. late) effects.
All statistical comparisons were made using Fisher's exact, two-tailed tests of the proportions of participants positive for a reaction, adverse event, or immune response, unless otherwise stated. The comparisons of reactogenicity and adverse events were based on the maximum severity per participant recorded at clinic visits. All analyses were performed using the SAS statistical software package.
Results
Study population
In total, 164 individuals were screened (Uganda: n = 46; South Africa: n = 71; and Zambia: n = 47) and 91 (45 males, 46 females) were enrolled [Uganda: n = 27; South Africa: n = 48 (16 per site); and Zambia: n = 16] between October 2005 and July 2006. Of these, 84 were randomized into Groups A–F to receive two intramuscular injections of 3 × 1010 (low dosage, LD, n = 22), 3 × 1011 (mid dosage, MD, n = 21), or 3 × 1012 (high dosage, HD, n = 21) DRP or placebo (n = 20) at intervals of either 6 months (early boost) or 12 months (late boost). Seven participants were enrolled into Group G in Uganda and were randomized to receive two intramuscular injections of 3 × 1012 DRP (n = 5) or placebo (n = 2) at the 6-month interval. Once the total number of individuals with anti-AAV2 neutralizing titers ≤1/8 enrolled in the protocol (including those chosen for Group G and those incidentally enrolled with low titers in other groups) reached 13, enrollment was halted. Because the results for Group G were similar to the corresponding HD recipients (Groups E and F) and corresponding placebo recipients (Groups A–F), safety and immunogenicity data of Group G were combined with the corresponding groups. Table 2 shows the characteristics of participants enrolled.
There were no statistically significant differences between dosage groups using Fisher's exact test (for gender and ethnicity) or Wilcoxon rank sum tests (for age and baseline AAV2 neutralizing titer).
Participants prescreened to have AAV2 neutralizing titers ≤1/8 (Group G, five vaccine, two placebo) were combined with corresponding recipients of 3 × 1012 DRP (Groups E and F) or placebo recipients (Groups A–F combined), since with the exception of baseline AAV2 neutralizing titers, the groups were similar.
Participants in early and late boost schedules combined by dosage group.
Eighty-two participants completed all vaccinations and study visits on schedule. Nine participants (two placebo, five LD, one MD, and one HD) did not receive the second vaccination due to pregnancy (n = 3), intercurrent HIV infection (n = 2), death (n = 1), preexisting undiagnosed medical illness (n = 1), investigator decision (n = 1), or relocation (n = 1). Of the nine participants who did not receive the second vaccination, six completed the protocol. There were no discontinuations of vaccinations or terminations due to vaccine-related adverse events.
Vaccine safety
Solicited adverse events
The rates of solicited local reactions and systemic signs and symptoms within 14 days of vaccination were similar among both vaccine and placebo recipients. Neither the severity nor the frequency of events was related to the dosage level. (Table 3). Approximately one-half to two-thirds of participants reported local reactions, regardless of their treatment assignment. Pain and/or tenderness at the injection site were the most common local reactions. All local events were mild except for one event of moderate pain in a placebo recipient. Approximately 50–75% of participants reported systemic signs and symptoms, none of which was severe. Seven vaccine recipients (three LD, two MD, and two HD) and one placebo recipient reported moderate systemic events, the most common being headache and fatigue. All other solicited systemic events were mild. The proportion of participants with local and systemic events in the 14 days following the first injection was statistically significantly higher than after the second injection in both vaccine and placebo recipients (p = 0.0002 for local reactogenicity, p = 0.0003 for systemic events; McNemar's test).
Data for subjects prescreened to have AAV2 neutralizing titers ≤1/8 (Group G, five vaccine and two placebo) were combined with corresponding recipients of 3 × 1012 DRP (Groups E and F) or placebo recipients (Groups A–F combined), since the results were similar.
Unsolicited adverse events
In total, 388 nonserious adverse events (AEs) were reported. Most AEs were mild (84%) or moderate (14%) in severity. The percentage of participants with AEs was similar between vaccine and placebo recipients [95%, 86%, 90%, and 96% for placebo, LD, MD, and HD, respectively (p = 0.60)]. The frequency and severity of AE's were not dose related and were similar between vaccine and placebo recipients during the 18-month follow-up period, as well as when the analysis was limited to AEs occurring within 28 days of vaccination (data not shown). Ten of 388 AEs (2.5%; eight mild, two moderate) were considered possibly related to the study product; none was judged probably or definitely related. The related AEs in vaccine recipients (n = 3) were classified under gastrointestinal disorders (abdominal cramps), infections and infestations (flu-like syndrome), and nervous system disorders (dizziness). AEs judged related in placebo recipients before unblinding (n = 7) were classified under infections and infestations (rhinitis), nervous system disorders (dizziness × 2), psychiatric disorder (insomnia), investigations (elevated ALT and elevated AST), and musculoskeletal and connective tissue disorder (intermittent muscle cramps).
Serious adverse events
Five serious adverse events were reported during the entire study, none of which was considered related to study product. All occurred prior to the second injection, one in a placebo recipient and two in LD and two in HD vaccine recipients. Four serious adverse events were hospitalizations for (1) malaria, (2) preeclampsia, (3) inguinal hernia, and (4) anemia due to bleeding duodenal ulcer. There was one death with a clinical diagnosis of viral encephalitis, with onset 4.5 months post vaccination (LD group). No etiology could be determined. PCR of the cerebrospinal fluid did not detect Herpes simplex types 1 and 2, enteroviruses (coxsackie A, B, echovirus), Epstein–Barr virus, mumps, and measles. Vaccine DNA was not detected in brain, liver, kidney, or reproductive tissues using a DNA-PCR assay (Targeted Genetics Corporation, Seattle, WA; limit of detection 15 copies/μg DNA). Pathologic findings of focal encephalitis were consistent with viral encephalitis. After a review of the comprehensive clinical and pathology data, the Safety Review Board agreed the event was unrelated to the study vaccine. It is noteworthy that other cases of presumed viral encephalitis, without an identified agent, occurred in the region at a similar time (L. Webber, personal communication).
Intercurrent HIV infection
Five study participants [two females, three males; 1/22 (5%) placebo and 4/69 (6%) vaccine recipients] acquired HIV infection during the 18-month study period due to exposure in the community. Vaccinations were complete in three participants and discontinued in two participants. Baseline anti-AAV2 capsid neutralizing titers for these participants ranged from 1/16 to 1/256. There was no evidence that acquisition of HIV infection was associated with baseline AAV2 neutralizing antibody titer >1/8 (p = 0.32) or assignment to vaccine versus placebo (p = 1.00). None of the vaccinees had developed positive ELISPOT responses against HIV-1 antigens prior to acquiring HIV infection. Viral loads at the time of diagnosis ranged between 261 and 81,000 copies/ml.
Pregnancies
Six women became pregnant during the study: three in the placebo group, two in the LD group, and one in the MD group. Three women conceived between the first and second vaccination and three conceived more than 4 months after the second vaccination (which was allowed per protocol). Five children were delivered at full term and one at 32 weeks of gestation. No congenital abnormalities were found.
Laboratory abnormalities
There was no consistent pattern of abnormal safety laboratory parameters. Most laboratory abnormalities were isolated, mild, judged to be clinically not significant, and resolved spontaneously. The most common abnormalities were neutropenia, thrombopenia, and elevated total bilirubin. Three grade 3 (severe) laboratory abnormalities were reported: (1) a grade 3 CD4 cell count (193 cells/mm3) in a male participant with preexisting insulin-dependent diabetes mellitus at the final study visit. Additional follow-up was arranged; 1 month later, the CD4 count was 270 cells/mm3 (moderate severity) and the HIV test was negative; (2) a grade 3 (severe) neutropenia (360 cells/mm3) in an acutely HIV-infected male; and (3) a grade 3 hemoglobin (and microcytic, hypochromic anemia) on the day of first HD tgAAC09 vaccination in a female participant who had normal hemoglobin at enrollment; a bleeding duodenal ulcer was diagnosed by gastroduodenoscopy. She was treated for Helicobacter pylorii and was hospitalized and received intravenous iron, but her hemoglobin remained grade 3 until the final study visit. She did not receive the second vaccination.
Neutralizing antibodies to AAV2
Baseline neutralizing antibody titers to the AAV2 capsid ranged from <1/4 to 1/2048, with a geometric mean titer (GMT) of 1/45. There were no differences in baseline AAV2 neutralizing titers between dosage groups (Table 2). The proportion of participants with moderate or greater solicited or unsolicited adverse events did not differ significantly between groups with lower (≤1/8) or higher (>1/8) anti-AAV2 neutralizing titers at baseline (p = 0.25 for all vaccine groups combined; p = 0.62 for the placebo group). The percentage of participants with a 4-fold or greater rise in neutralizing antibody titers after vaccination increased with dosage of tgAAC09; it was 59%, 90%, and 96% for LD, MD, and HD, respectively, and 23% for placebo recipients (Cochran–Armitage trend test; p < 0.0001). The increase of AAV2 antibody titers in placebo recipients may be due to naturally circulating AAV in the community.
Vaccine shedding
Biodistribution specimens from four HD and two placebo recipients were assayed in a blinded fashion. There was no evidence of vaccine shedding in nasal secretions, saliva, urine, semen, or PBMCs after intramuscular doses of tgAAC09 at 3 × 1012 DRP. One day 14, a cervicovaginal secretion sample from a placebo recipient, was positive with a faint band on the PCR readout. There were several inconclusive tests in the same assay, suggesting that there was inhibition by the cervicovaginal sample matrix. This result was considered a false positive, because the participant received placebo. Of the 147 specimens collected during the study, no other specimens were positive.
Vaccine immunogenicity
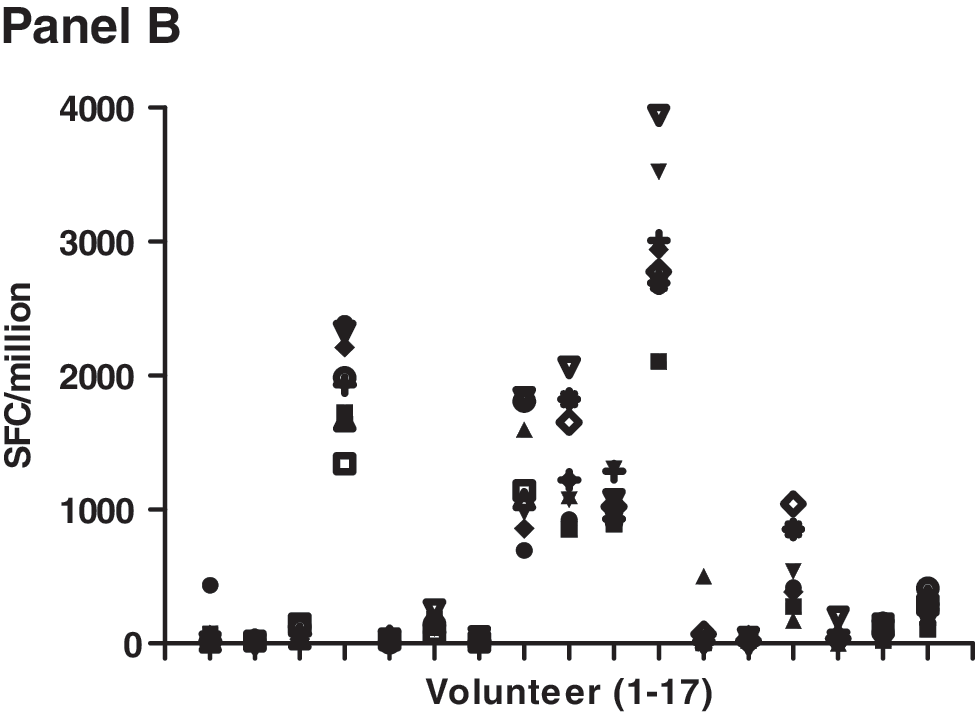
Cellular immune responses were dose related (Table 4). Overall response rates were 0% for the placebo recipients, 9% for LD, 24% for MD, and 38% for HD vaccinees (Cochran–Armitage trend test; p = 0.0001). Most responses were to Gag and the magnitude was generally modest as shown in Fig. 1 (range: 42–1455; median: 64; mean: 141 SFC/106 PBMCs). The rate of responders to at least one of the Gag pools A–D was similar to the rate of response to the whole Gag pool 120; this is illustrated by an individual in the LD group who had an equivalent response to both the Gag pool B and the whole Gag pool at 2 months after the first immunization (Fig. 1, LD panel). The duration of the ELISPOT response was limited, but appeared to improve at the high dosage. Two participants responded to protease Pool 7 (one each in the MD and HD group). Eight vaccine recipients (one LD, one MD, and six HD) responded to at least two Gag peptide pools. ELISPOT responses were seen sporadically over the time course of vaccination; six vaccine recipients (one MD, five HD) responded on more than one occasion to Gag. In the MD and HD groups, the ELISPOT response rate was significantly increased with boosting at 6 months (p = 0.01; McNemar's test), whereas boosting at 12 months produced no new ELISPOT responses. ELISPOT responses to FEC peptides were as expected with consistent responses across visits. This is illustrated in Fig. 1B for 17 LD, MD, and HD individuals who showed HIV-specific responses. Across the range of FEC responses (0 to several thousand SFC/million PBMCs) the magnitude for each individual was similar across visits indicating that there was consistency in the PBMC sampling and ELISPOT assays across visits in spite of low HIV-specific responses. In addition, it should be noted that PHA responses were robust across visits for all groups (data not shown). Because of the sporadic nature and low magnitude of the ELISPOT responses as well as the paucity of anti-HIV-specific antibodies, no intracellular cytokine assays were performed to determine whether the responses were mediated by CD4 or CD8 T cells.
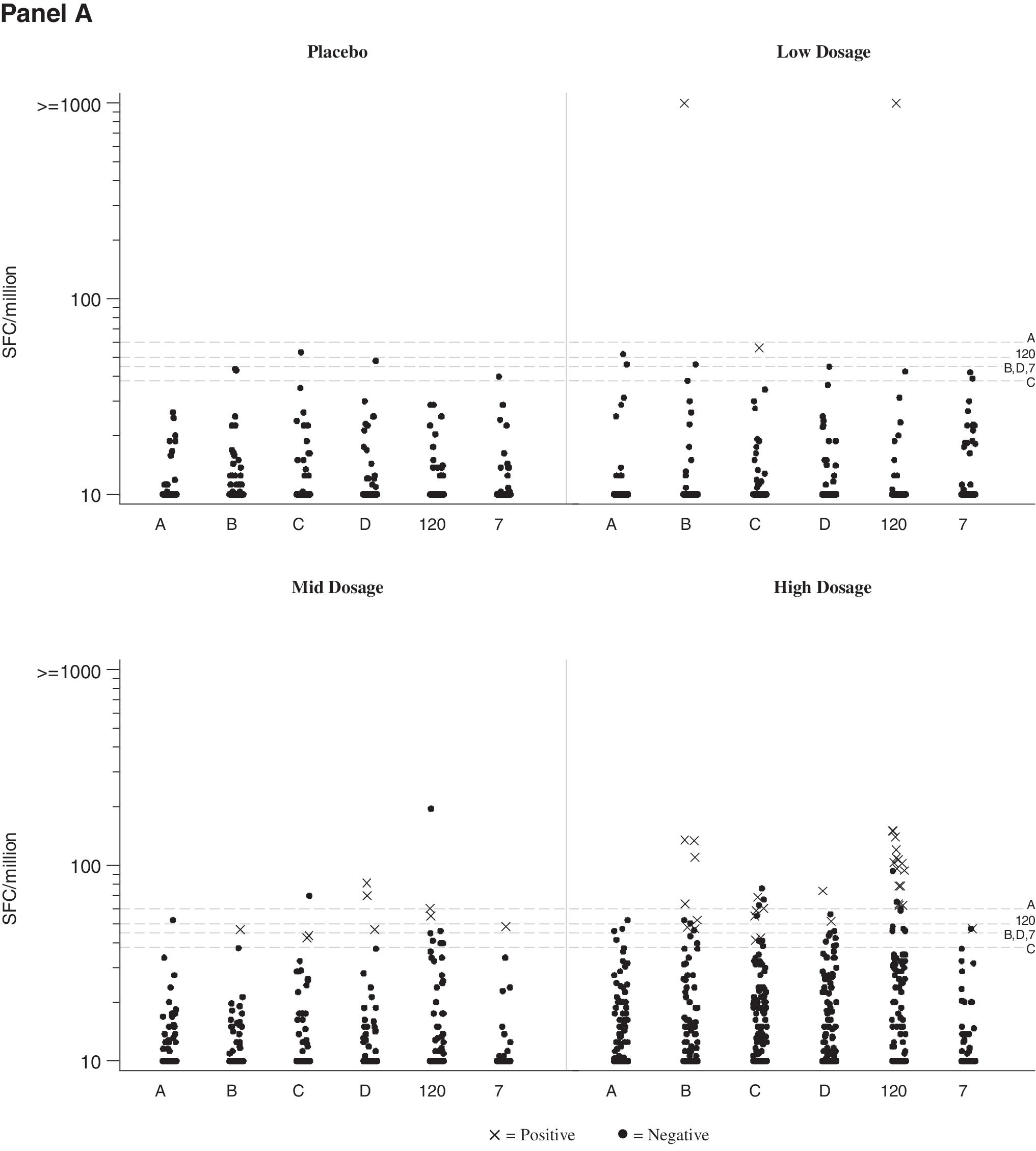
IFN-γ ELISPOT responses.
Data for subjects prescreened to have AAV2 neutralizing titers ≤1/8 (Group G, five vaccine, two placebo) were combined with corresponding recipients of 3 × 1012 DRP in Group E (early boost) or placebo recipients (Groups A, C, E combined) group, since the results were similar.
Although the numbers are small, the presence of AAV2 neutralizing titers at baseline did not appear to adversely impact the development of T cell immunity to HIV vaccine antigens in any dosage group. Among participants who received two doses of tgAAC09 of 3 × 1012 DRP 6 months apart, T cell responses were seen in 4/9 (44%) vaccinees with baseline AAV2 neutralizing titers ≤1/8 compared to 3/7 (43%) vaccines with baseline AAV2 neutralizing titers >1/8 (baseline GMT 1:116, range 1/16 to 1/1024). In a post-hoc analysis of all vaccine recipients, 6/17 (35%) with baseline titers ≤1/8 and 11/52 (21%) with baseline titers >1/8 responded to the vaccine (p = 0.67), adjusting for dosage and vaccination schedule.
Anti-Gag antibodies were detected in three vaccinees at one time point each in one LD recipient 6 months after the first injection and two HD recipients 2 months after the boost at 12 months. The two HD ELISA responders also had positive ELISPOT responses in the trial at multiple time points (data not shown).
At the final study visit, no vaccine recipient had a positive HIV test results due to vaccine-induced antibodies.
Discussion
In a previous study, a single administration of tgAAC09 was well tolerated after intramuscular injection at three dosage levels up to 3 × 1011 DRP. In this study, the vaccine candidate was well tolerated after two intramuscular injections at dosages up to 3 × 1012 DRP. The rates of adverse events, including solicited local and systemic events within 14 days of vaccination, were not dose related and were similar in frequency and severity between vaccine and placebo recipients. After the first injection, almost all of the solicited events were reported more frequently than after the second injection, but this was true of placebo as well as vaccine recipients; it may be due to a reporting bias. The rates were also higher than reported in the previous clinical trial. 6 The reasons for this are unclear, but perhaps there are differences in underlying health status or perceptions between African participants and those in Europe or India.
No published data on preexisting anti-AAV2 capsid neutralizing titers in African populations exist, but unpublished data from a serosurvey conducted by IAVI suggest that the prevalence of AAV2 antibodies is approximately 75%. Preexisting immunity to AAV2 did not appear to affect reactogenicity.
All mild and some moderate abnormalities of total bilirubin, absolute neutrophil count, and low hemoglobin occurring in Ugandan and Zambian volunteers were within consensus ranges established in healthy African adult males and females. 11 The respective values would have been considered normal had consensus ranges been used as reference by the respective local clinical laboratories.
In nonhuman primates, a recombinant AAV-based analogue SIV vaccine induced long-lasting cellular immunity and antibodies to the expressed proteins and provided partial protection. The strong T cell and antibody-mediated immune responses observed in macaques were not reproduced in this clinical study; overall the magnitudes of the ELISPOT and antibody responses were low and sporadic. HIV gag-specific T cell responses are infrequently seen in response to vaccination and since only gag and protease responses were examined it is unknown whether a different immunogen in the AAV2 vector might induce more robust responses.
As suggested in the previous clinical study, 6 a dose-response relationship for immunogenicity was also observed in this study. In the two higher dosage groups, the ELISPOT response rate significantly improved with boosting at 6 months, but not at 12 months, suggesting that immune memory is transient and wanes between 6 and 12 months. The results of this study do support the hypotheses that a higher dosage was needed, but they do not provide strong support for the hypothesis that a booster dose of the same vaccine would greatly increase the responses.
Preexisting immunity to the AAV2 vector was of concern; so few participants had low or no AAV2 neutralizing titers at baseline that it was difficult to determine the effect of preexisting immunity to the AAV2 capsid on the immune response to HIV antigens encoded by the vector. However, the response rate in the HD group for participants with low or no prevaccination AAV2 neutralizing titers was similar to those with higher AAV2 neutralizing titers.
Vaccine recipients made robust, dose-related antibody responses to the AAV2 capsid protein; these responses are to the injected vector, as no AAV2 capsid gene is present in the vaccine, whereas they responded only modestly to the HIV Gag protein, which was probably expressed over days to weeks postinjection. The reasons for the modest immunogenicity of tgAAC09 in humans are unclear, but may relate to the low level of inflammation associated with the AAV2 vector, as observed by the similar reactogenicity in both vaccine and placebo recipients. The quantity of vaccine expressed may be a limiting factor. Increased dosage, increased expression per vector particle, and/or a combined regimen of AAV2 with a different vector may improve immune responses. In this study, the dosage of tgAAC09 was increased to the highest practical level (3 × 1012 DRP).
Despite entry criteria to exclude participants at risk for HIV infection and repeated risk-reduction counseling, five incident infections were detected during this 18-month study; the distribution between vaccine and placebo recipients was proportional to the randomization to vaccine and placebo. The limited benefit of risk reduction counseling in HIV prevention underscores the need to develop an effective HIV vaccine.
Further clinical testing of this HIV vaccine candidate as a single agent is not warranted. However, recombinant AAV2 vectors containing more or different immunogens may be a useful prime for a different vector-based vaccine.
Footnotes
Acknowledgments
We express our thanks to all the study participants, to the research staff at the collaborating research centers (PHRU: Simikiwe Mayaphi, Tebogo Magopane, Mabela Molapo-Matsoso, Anusha Makuraj; UVRI-IAVI: Annet Nanvubya, Fiona Kalinda, Paul Kato Kitandwe, Aloysius Ssemaganda, Enoch Muyanja, AnneMarie Namuniina; Cape Town: Keren Middelkoop, Roland Croxford, Joan Aploon; Medunsa: Maposhane Nchabeleng, Samukeliso Dube, Innocentia Lehobve, Yusuf Dangor; Zambia: Cheswa Vwalika, William Kilembe, Olivier Manigart) for their outstanding work and dedication, and to the Safety Review Board for overseeing the study. We also extend our thanks to the IAVI Clinical Program Managers (Helen Thomson, James Sherwood, Bruce Johnson, Jennifer Lehrman, and Dani Vooijs), Eddy Sayeed (Vaccine development, IAVI, NY), and Jim Ackland (regulatory consultant). This study was funded by the International AIDS Vaccine Initiative (IAVI). IAVI's financial and in-kind supporters include the Alfred P. Sloan Foundation, the Bill & Melinda Gates Foundation, The John D. Evans Foundation, The New York Community Trust, the James B. Pendleton Charitable Trust, The Rockefeller Foundation, The Starr Foundation, The William and Flora Hewlett Foundation; the Governments of Canada, Denmark, Ireland, The Netherlands, Norway, Sweden, the United Kingdom, and the United States, the Basque Autonomous Government as well as the European Union; multilateral organizations such as The World Bank; corporate donors including BD (Becton, Dickinson & Co.), Continental Airlines, Google Inc., Merck & Co., Inc., and Pfizer Inc; leading AIDS charities such as Broadway Cares/Equity Fights AIDS and Until There's A Cure Foundation; other private donors such as The Haas Trusts; and many generous individuals from around the world. This work was made possible with funding from the International AIDS Vaccine Initiative, including funding from USAID Cooperative Agreement Number GPO-A-00-06-00006-00. The contents of this manuscript are the responsibility of IAVI and do not necessarily reflect the views of USAID or the U.S. government. For more information, see
Author Disclosure Statement
P.R.J. is named on a patent application for this vaccine concept. P.A. and A.E.H participated in this study as employees of Targeted Genetics Corporation. M.B., J.L, C.S., J.G., and P.F. participated in this study as employees of the International AIDS Vaccine Initiative (IAVI).