
Abstract
An HIV-1 subtype C specific assay was established for integrase genotyping from 51 integrase inhibitor-naive patient plasma samples and 22 antiretroviral drug-naive primary viral isolates from South Africa. Seventy-one of the 73 samples were classified as HIV-1 subtype C and two samples were unique AC and CG recombinants in integrase. Amino acid sequence analysis revealed there were no primary mutations (Y143R/C/H, Q148H/R/K, and N155H/S) associated with reduced susceptibility to the integrase inhibitors raltegravir and elvitegravir. However, one sample had the T97A mutation, three samples had the E157Q and V165I mutations, and the majority of samples contained the polymorphic mutation V72I. The expected finding of no major integrase mutations conferring resistance to integrase inhibitors suggests that this new antiretroviral drug class will be effective in our region where HIV-1 subtype C predominates. However, the impact of E157Q and other naturally occurring polymorphisms warrants further phenotypic investigation.
T
Integrase functions in cells within the context of high-molecular-weight preintegration complexes that are formed in the cytoplasm after reverse transcription of the viral RNA. HIV-1 integration is subdivided into two steps with distinct kinetics and requirements. In the cytoplasm, a dinucleotide adjacent to a phylogenetically conserved CA is hydrolytically cleaved off the 3′ end of each strand of the linear viral cDNA by integrase in the preintegration complex (3′-processing). 3 –7 After nuclear transport of the preintegration complex, two phosphodiester bonds on each strand of the chromosomal DNA are targeted by the 3′-hydroxyls at the ends of the viral DNA, resulting in its insertion within the host genome (strand transfer). 3 –7 Removal of the two unpaired nucleotides on the 5′ viral DNA ends and gap filling of the interfaces between the viral and host genomic DNA are then done using host DNA repair machinery via mechanisms that are not yet fully understood. Completion of integration results in a fully functional provirus, which can then be used to initiate viral DNA transcription. There is no known human homologue to integrase, thus making it an ideal antiretroviral drug target.
Raltegravir (Isentress, Merck & Co., Inc., Whitehouse Station, NJ), the first integrase inhibitor to be approved by the U.S. Food and Drug Administration (October 2007), has successfully been used in combination with other antiretroviral drugs for treatment-experienced HIV-1 patients with multidrug-resistant strains. Raltegravir blocks viral replication by inhibiting DNA strand transfer.
8
However, drug resistance to integrase inhibitors has been shown to occur both in vivo and in vitro, and is the consequence of mutations that are selected in the viral integrase gene targeted by the strand transfer antiretroviral drugs such as raltegravir and elvitegravir (Gilead Sciences, Foster City, CA). To date, there are at least 30 substitutions that have been specifically associated with the development of resistance to raltegravir and/or elvitegravir (
Access to antiretroviral drugs has escalated dramatically in the sub-Saharan African region in the past decade. Regimens in this region almost universally place a thymidine analogue with lamivudine and a nonnucleoside reverse transcriptase inhibitor as first line therapy, with a protease inhibitor-based regimen for those failing first line therapies. However, these regimens carry significant toxicities, requiring drug substitutions. In addition, an increasing number of patients require alternative regimens as they fail first and second line regimens due to nonadherence, toxicity, or the development of drug resistance. Thus, the effectiveness of integrase inhibitors such as raltegravir needs to be investigated as alternative antiretroviral drug treatment options are required. Several groups have published genotypic assays for the amplification and sequencing of integrase from diverse HIV-1 group M subtypes, 9 –11 however, we opted to establish a subtype C-specific integrase genotyping assay. This study describes the baseline mutations in integrase in HIV-1 subtype C (and unique recombinant) infected integrase inhibitor-naive individuals in South Africa.
A total of 80 samples from patients attending the Charlotte Maxeke Johannesburg Hospital were available for purposes of this study. These included 58 randomly selected patient plasma samples sent for routine viral load monitoring during 2009, and 22 well-characterized primary HIV-1 isolates from antiretroviral drug-naive AIDS patients isolated during 2005. 12 The patients viral loads ranged from 1500 to >3 million RNA copies/ml, and included antiretroviral drug-naive and treatment-experienced patients. Ethical clearance for the study was obtained for Research on Human Subjects (Medical) at the University of the Witwatersrand (clearance number M090575). Viral RNA was extracted from the 58 patient plasma samples using the automated Roche MagNa Pure LC analyzer and the MagNa Pure LC Total Nucleic Acid Isolation kit (Roche, Germany), according to the manufacturer's instructions. Proviral DNA from the 22 viral isolates was extracted from p24 antigen-positive cultures with a High Pure PCR Template Preparation kit (Roche, GmbH, Germany) according to the manufacturer's instructions. The full-length integrase gene was reverse transcriptase polymerase chain reaction (RT-PCR) amplified into cDNA from the extracted viral RNA samples using the reverse primer SQ9RC(3) (5′-TTGGATGTCTGCTTTCATAATGAT-3′) 13 and the Expand Reverse Transcriptase (RT) kit (Roche Diagnostics, Germany), as per the manufacturer's instructions. Amplification from synthesized cDNA or extracted proviral DNA was performed using primers SQ7F(2) (5′-AGGAGCAGAAACTTTCTATGTAGATGG-3′) 13 and SQ9RC(3) with the Expand High FidelityPLUSkit (Roche, Germany), as per the manufacturer's instructions. PCR amplicons (approximately 1.3 kb in length) were purified using the High Pure PCR Product Purification kit (Roche), as per the manufacturer's instructions.
Primers spanning the full-length integrase (∼880 bp) were used to sequence the PCR products in both directions (population-based sequencing). These included QFIN1F (5′- ATGGGTACCAGCACACAAAG-3′), QFIN2F (5′-TAGCAGGAAGATGGCCAGTC-3′), QFIN1R (5′-CTCTCCTTGAAATATACATATGGTGC-3′), and QFIN2R (5′-CTACTACTCCTTGACTTTGGGG-3′). Purified amplicons were sequenced with the ABI Big Dye terminator system (v3.1) according to the manufacturer's instructions on the ABI Prism 3730 and 3100-Avant Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequence data were edited using the Sequencing Analysis V3.3 program (Applied Biosystems), and the complete integrase sequences were assembled and manually edited using Sequencher V4.7 (Genecodes, Ann Arbor, MI). For subtyping, a multiple alignment of the full length integrase region with references from HIV-1 subtypes A to K, CRF01_AE, and CRF02_AG (
RT-PCR and PCR amplification was successful for 91.3% of all samples, and included 51 of the 58 patient plasma samples (not dependent on viral load) and all 22 proviral DNA samples, respectively. All 73 of the amplified samples were successfully sequenced. The integrase amplification and sequencing success rates are similar to those reported previously.
9
–11
Nucleotide sequences of the 73 newly characterized full-length integrase genes ranged from 867 to 876 base pairs, with intact open reading frames confirming the presence of functional integrase genes in all patients and primary virus isolates. The integrase intrasubtype nucleotide diversity for all 73 newly characterized samples varied between 2.2% and 7.9%, similar to what has been reported previously.
14
Phylogenetic analysis of the full-length integrase nucleotide sequences, together with viruses from the major subtypes, revealed that 71 sequences clustered within HIV-1 subtype C with a bootstrap value of 100%, whereas two samples (09ZAP35 and 09ZAP52) showed evidence of recombination (Fig. 1). These two sequences were further analyzed using the RIP 3.0 Program (
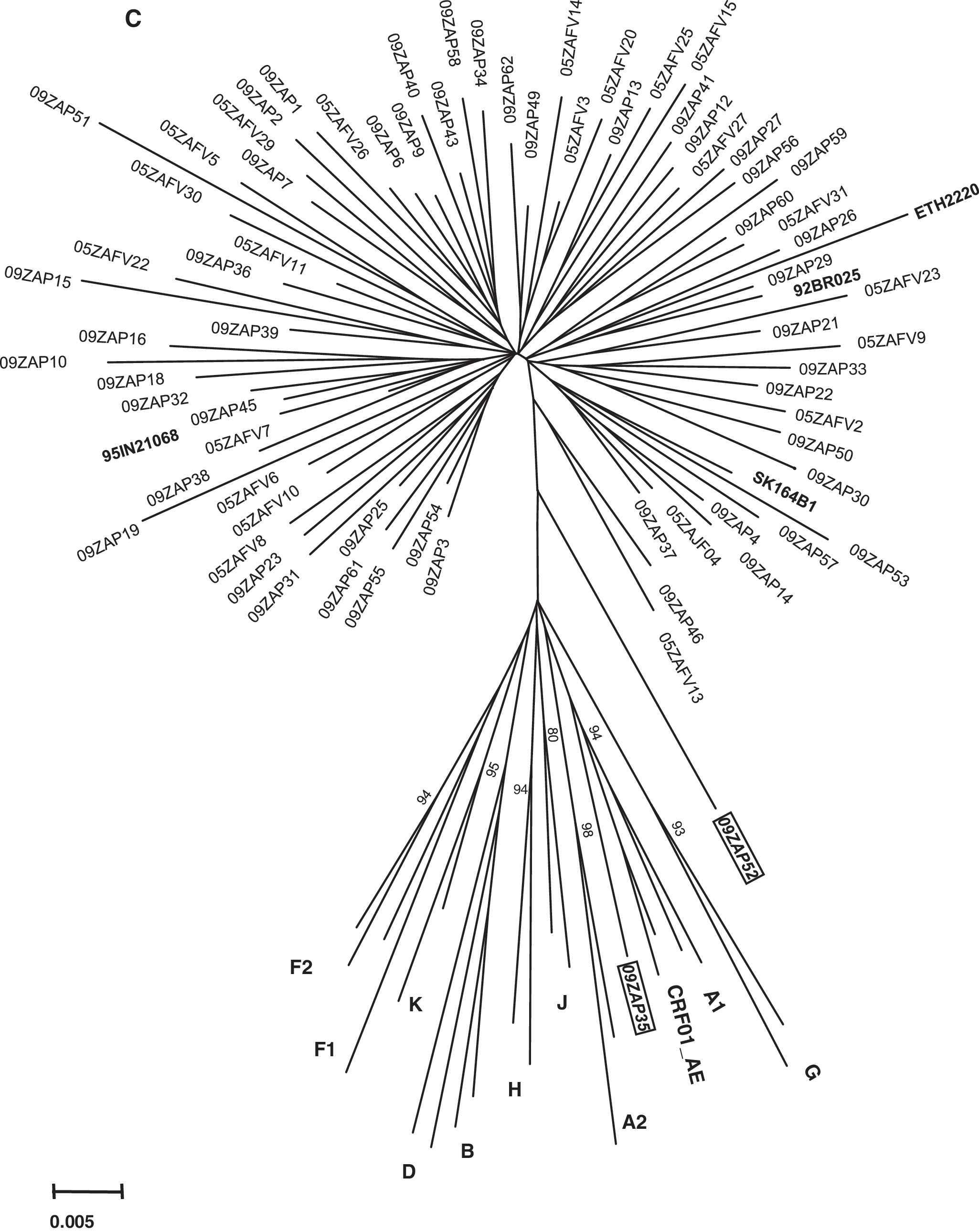
Phylogenetic relationships of the 73 newly characterized full-length integrase sequences with HIV-1 subtype reference sequences from the Los Alamos database (
An alignment of the 73 integrase amino acid sequences was extensively analyzed and compared, and a distribution of the variants is shown in Fig. 2. The proteins varied from 288 to 292 amino acids in length, primarily due to the presence of an unusual tetrapeptide insertion (QNME) in the C terminus of the protein in 23 patient and primary virus isolate samples. This unusual insertion has been noted previously. 15,16 The C terminus has been shown to be important for integrase dimerization and DNA binding. This is crucial for proper function and positioning of the catalytic domain close to its target site. This finding warrants further investigation to determine the impact of the insertion on the function of the integrase enzyme.

Distribution of variants among the 73 newly characterized full-length integrase sequences. The 2002 consensus subtype C sequence (
The sequences were analyzed for the presence of integrase mutations associated with reduced in vitro susceptibility to raltegravir (E92Q, F121Y, E138AK, G140AS, Y143RCH, S147G, Q148HRK, N155HS) and elvitegravir (T66IAK, E92Q, F121Y, E138AK, G140AS, S147G, Q148HRK, S153Y, N155HS, R263K) (Stanford Database, updated 15 September, 2009). None of the samples contained the mutations associated with primary resistance (Y143RCH, Q148HRK, and N155HS).
The E157Q mutation was observed among 4.1% (n = 3) of patient samples, including two subtype C patient samples (09ZAP34 and 09ZAP53) and one recombinant (09ZAP52) patient sample (Fig. 2). Until recently the E157Q mutation was listed among the mutations associated with reduced in vitro susceptibility to raltegravir and elvitegravir (
The isoleucine at position 72 (I72) was identified at a high frequency among the subtype C samples and the 05ZAP52 recombinant (67.1%; n = 49), whereas V72 and T72 were found in 28.7% (n = 21, including the 05ZAP35 recombinant) and 1.4% (n = 1) of the patients, respectively, confirming their status as a naturally occurring subtype C polymorphism. This confirms the findings by Rhee et al. 14 who characterized naturally occurring variation in previously published HIV-1 group M integrase sequences, and found that 390 of 432 (90.3%) integrase-inhibitor-naive subtype C samples analyzed contained isoleucine at position 72, and only 9.4% and 0.2% contained a valine and threonine, respectively. The polymorphic mutation, T97A, associated with integrase inhibitors was observed in one primary virus sample (05ZAFV27), whereas K156N, another accessory integrase inhibitor resistance mutation, was found in patient 09ZAP34. The uncommon polymorphism K156N was not reported in subtype C, but occurs in 9% of subtype B and 5% of subtype D sequences. 14 By contrast, T97A was observed in subtypes A, B, C, D, and CRF01_AE and CRF02_AG, albeit at low frequencies. 14
Because the antiretroviral drug history of the patient samples analyzed in 2009 was unknown, we could not address the influence of prior antiretroviral therapy on integrase polymorphisms. Ceccherini-Silberstein et al. 18 reported several integrase polymorphisms, for example, M154I, V165I, and M185L, that were positively associated with specific reverse transcriptase mutations (F227L and T215Y) in treated patients. In our study, the V165I polymorphism was noted in three samples, 09ZAP3, 09ZAP54 (unknown drug history), and 05ZAFV29 (antiretroviral drug naive). This highlights the need for ongoing surveillance of integrase mutations as well as HIV-1 subtype C-specific phenotypic studies to elucidate the relevance of the above-mentioned polymorphisms (as well as naturally occurring polymorphisms) in the absence of the signature integrase mutations on susceptibility/resistance to integrase inhibitors.
In conclusion, none of the three previously reported major mutations (Y143R/C/H, Q148H, and N155H) associated with resistance to integrase inhibitors was observed in our cohort, indicating that integrase inhibitors may be used as a treatment option in South Africa.
Sequence Data
The 73 integrase sequences were submitted to GenBank using Sequin V5.35 (
Footnotes
Acknowledgments
We are grateful for research funding from AuTEK Biomed (Mintek and Harmony Gold Mining Company) and the Department of Molecular Medicine and Haematology.
Author Disclosure Statement
No competing financial interests exist.