
Abstract
We determined the incidence of and risk factors for distal sensory polyneuropathy (DSP) in individuals on HAART. Sixty-one HIV-positive subjects on HAART for at least 6 months and neuropathy free were retrospectively selected. The study included subjects who had previously tolerated d-drugs without developing DSP. Neuropathy incidence over 4 years was calculated. Cox proportional hazards models were used to determine risk factors associated with incident DSP. Nineteen subjects developed DSP over a mean follow-up of 2.4 years. Subjects never treated with a d-drug developed DSP at a rate of 21 cases per 100 person-years (95% CI, 8.9–33.7). Subjects with a history of d-drug treatment but not on a d-drug at enrollment developed DSP at a rate of 17 cases per 100 person-years (95% CI, 2.1–31.8). Those on d-drug treatment developed DSP at a rate of 25 cases per 100 person-years (95% CI, 8.7–41.6). Multivariable analysis identified age [hazard ratio (HR) = 1.09; p < 0.01] and low CD4+ nadir [hazard ratio (HR) = 0.79; p = 0.03] as significant risk factors. Current or prior history of treatment with d-drug was not a significant risk factor for incident DSP in subjects who had previously tolerated d-drug treatment without developing a toxic DSP. Age and low CD4+ are risk factors for incident DSP. However, current or prior history of d-drug treatment is not a significant risk factor for incident DSP in subjects who had previously tolerated d-drug treatment without developing a toxic DSP.
Introduction
S
Prior to the use of HAART, HIV-associated DSP was associated with markers of advanced immunosuppression (e.g., high plasma HIV viral load, reduced CD4+ lymphocyte cell count), 7 affecting approximately one-third of infected patients. 5 In the early HAART era, three dideoxynucleoside reverse transcriptase inhibitors (d-NRTI)—zalcitabine (Hivid, 2′,3′-dideoxycytidine or ddC), stavudine (Zerit, 2′,3′-didehydro-3′-deoxythymidine or d4T), didanosine (Videx, dideoxyinosine or ddI)—known collectively as “d-drugs” caused toxic DSP, which was clinically difficult to differentiate from HIV-associated DSP. 8 –11
The risk of developing a toxic DSP from a d-drug usually peaks in the first 3 months of initiating therapy. 12 One study 13 suggested that if DSP did not develop within the first year of using a d-drug, it was less likely to occur in subsequent years. Nonetheless, it is still controversial whether exposure to d-drugs has a cumulative toxic effect and whether continued d-drug exposure in those who had previously tolerated these agents without neurotoxicity is associated with an increased risk of developing DSP. D-drugs were extensively used in the United States and Europe, and the issue of continued future risk associated with previous d-drug use is of particular relevance as the HIV-infected population ages. Moreover, as medications with less neuropathy risk replace the use of d-drugs in resource-limited countries, it is important to determine whether patients tolerating d-drugs necessarily need to switch to other HIV medications to avoid higher risk of neuropathy if they are doing well. This study therefore aimed to determine the incidence of and risk factors for DSP in patients on HAART with particular attention to assessing potential incident neuropathy risk in individuals who had previously tolerated treatment with a d-drug without developing DSP.
Materials and Methods
Study design
A retrospective convenience sample of subjects who were on HAART and without DSP at enrollment was selected from the Hawaii Aging with HIV Cohort (HAHC) (Fig. 1).
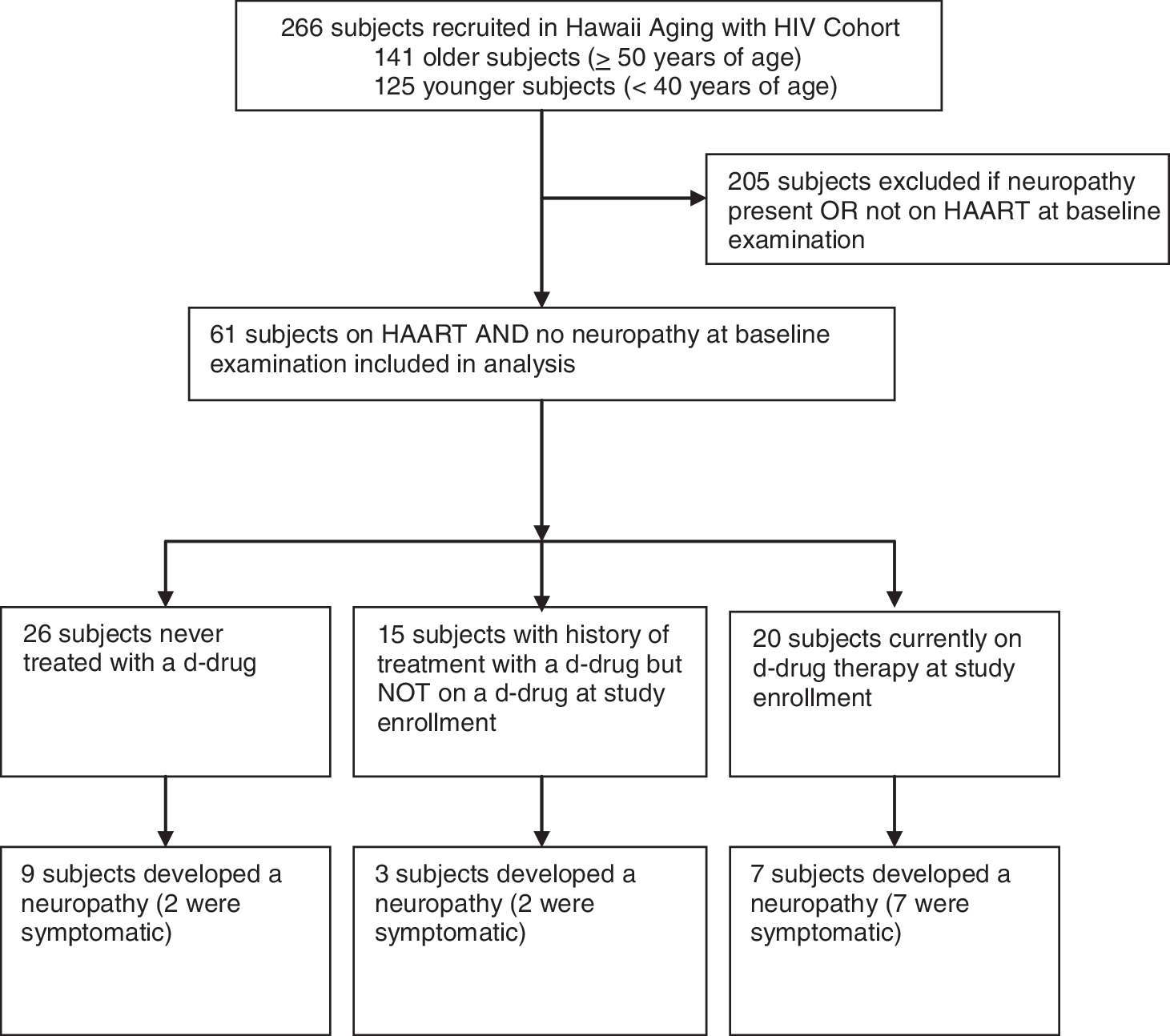
Flow chart of subject selection and outcome.
Hawaii aging with HIV cohort
The HAHC was a prospective study examining the relationship between aging and neurocognitive impairment in HIV-infected individuals. Details of enrollment and clinical characterization have been published elsewhere. 14
Briefly, 141 older (≥50 years of age) and 125 younger (<40 years of age) HIV-infected individuals residing in the state of Hawaii were enrolled from October 22, 2001 to July 1, 2005. Primary exclusion criteria included premorbid or comorbid conditions that might limit completion or interpretation of neuropsychiatric evaluations, including intravenous drug use, within 6 months of enrollment, active major psychiatric disorders, history of traumatic brain injury with loss of consciousness for more than 1 h, poorly controlled epilepsy, history of stroke, active CNS opportunistic disease, and learning disabilities. Baseline and annual evaluations included demographic data, medical and medication history, a neurologic examination, and HIV laboratory parameters (plasma HIV viral load, current CD4+ lymphocyte cell count, and self-reported lowest ever CD4+ lymphocyte cell count). All subjects signed informed consent, and the institutional review board at the University of Hawaii approved the study.
Neuropathy assessment
The HAHC study utilized a macroneurological examination for the assessment of neuropathy created by the AIDS Clinical Trials Group. 15 The examination included tests of sensation to pinprick, light touch, and vibration on all four extremities, comparing proximal (at or above the knee/elbow) to distal sensation (at or below the ankle/great toe/wrist/finger). Vibratory impairment was defined as less than 10 s of perception after a 128-Hz tuning fork was struck with maximal force and applied to the base of the great toe, ankle, knee, distal interphalangeal joint of the middle finger, or wrist. A semiquantitative assessment of severity based on level of neuropathic signs was performed. Sensation to each modality was documented as no impairment, impairment at or below the ankles/wrists, impairment between the ankle and knees/wrist and elbow, or impairment at or above the knees/elbows. Ankle reflexes were examined and compared to deep tendon reflexes at the knee. Diagnosis of a DSP required both diminished or absent ankle jerks reflexes compared to the knee and a diminution of sensation in at least one modality present in the lower extremities bilaterally. Symptomatic DSP required an examination consistent with DSP and the presence of two or more neuropathic symptoms, which included numbness, tingling, burning, stinging, hypersensitivity, or pain. Electrophysiologic studies, autonomic reflex tests, and skin biopsies were not performed.
Statistical analysis
Statistical analysis was performed with SAS 9.1 (Cary, NC). Means, standard deviations, and proportions were calculated for baseline characteristics. Incidence of DSP in 100 person-years was defined as the number of incident cases of neuropathy divided by the total duration of at-risk and multiplied by 100. A Cox proportional hazards model was used to test the null hypothesis that rate of progression to DSP is no different between subjects never treated with a d-drug, subjects currently on d-drug treatment, and subjects with a history of treatment with a d-drug but not on a d-drug at study enrollment. The presence or absence of DSP on neurological examination was the dependent variable. Based on the results of previous published studies, 5,16,17 age, CD4+ nadir, years of d-drug treatment, current HIV RNA viral load, and current CD4+ cell count were included as independent variables. Univariable analysis was performed, followed by a multivariable analysis that included independent variables with a p-value ≤ 0.1 in the univariable analysis.
Results
Subject characteristics
Sixty-one HIV-positive subjects without DSP and on HAART at enrollment were included in this analysis (Fig. 1). The demographics and clinical characteristics of these subjects are summarized in Table 1. Twenty-six subjects (43%) were never treated with d-drugs. Fifteen subjects (24%) had a history of treatment with a d-drug but were not on a d-drug at study enrollment. Twenty subjects (33%) were on d-drug treatment at enrollment. A majority of the subjects had undetectable HIV viral loads and CD4+ cell counts greater than 200 cells/μl. A minority of the subjects had current alcohol abuse defined by the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV), 18 neurotoxic drug exposure other than a d-drug (e.g., isoniazid, hydroxyurea, ethambutol), or other neurotoxic medical comorbidities (e.g., diabetes, impaired glucose tolerance, uremia, vitamin B12 deficiency, thyroid disorders, connective tissue disorders, syphilis, or solvent exposures).
Current alcohol abuse was defined by Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV).18
Other neurotoxic drug exposures were obtained by self-report and included isoniazid, hydroxyurea, and ethambutol.
Neurotoxic medical comorbidities were obtained by self-report/medical chart review and included diabetes, impaired glucose tolerance, uremia, vitamin B12 deficiency, thyroid disorders, connective tissue disorders, syphilis, and solvent exposure (e.g., carbon disulfide, n-hexane, methyl butyl ketone, perchloroethylene, trichloroethylene, toluene).
The sample contained two groups: 28 older (≥50 years of age) and 33 younger (<40 years of age) subjects. The older group were HIV seropositive longer (13 years, standard deviation 5.7 years) compared to the younger group (8 years, standard deviation 6.0 years, p < 0.01). There were no statistically significant differences between the older and younger groups in CD4+ nadir, proportion who had any d-drug exposure (i.e., history of d-drug treatment but not on a d-drug at enrollment or on d-drug treatment at enrollment), and proportion who developed incident symptomatic DSP.
Neuropathy incidence
Nineteen subjects developed a DSP over a mean follow-up of 2.4 years (Fig. 1). Subjects with no history of d-drug treatment developed a DSP at a rate of 21 cases per 100 person-years (95% CI, 8.9–33.7 cases per 100 person-years). Subjects with a history of d-drug treatment but not on a d-drug at enrollment developed a DSP at a rate of 17 cases per 100 person-years (95% CI, 2.1–31.8 cases per 100 person-years), whereas those on d-drugs developed a DSP at a rate of 25 cases per 100 person-years (95% CI, 8.7–41.6 cases per 100 person-years).
Neuropathy risk factors
Univariable analysis identified age and CD4+ nadir as significant risk factors (Table 2). These risk factors were included in a multivariable analysis. A subject's d-drug exposure history was also included in the model since this was a variable of interest. Subjects currently on a d-drug and those with history of d-drug treatment but not on a d-drug at enrollment were compared to subjects never treated with a d-drug.
Subjects never treated with a d-drug were used as the reference group.
Current alcohol abuse was defined as defined by Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV). 18
Other neurotoxic drug exposures were obtained by self-report and included isoniazid, hydroxyurea, and ethambutol.
Neurotoxic medical comorbidities were obtained by self-report/medical chart review and included diabetes, impaired glucose tolerance, uremia, vitamin B12 deficiency, thyroid disorders, connective tissue disorders, syphilis, and solvent exposure (e.g., carbon disulfide, n-hexane, methylbutyl-ketone, perchloroethylene, trichloroethylene, toluene).
Multivariable analysis identified age and CD4+ nadir as significant risk factors associated with incident DSP (Table 3). The incidence of DSP increased by 9% for every year of age [hazard ratio (HR) = 1.09; p < 0.01] and increased by 21% for every 100 cells/ml decrease in CD4+ nadir [hazard ratio (HR) = 0.79; p = 0.03]. Using subjects never treated with a d-drug as the reference group, subjects currently on a d-drug and those with prior d-drug exposure but not on a d-drug at enrollment were not significant risk factors associated with incident DSP.
Subjects never treated with a d-drug were used as the reference group. d-NRTI, dideoxynucleoside reverse transcriptase inhibitor.
Length of exposure to d-drug
Fifteen subjects had a history of d-drug treatment but were not on a d-drug at enrollment (Fig. 1). These subjects had previously been treated with d-drugs for a median of 1.0 years (interquartile range of 0.60–4.41 years); three developed a DSP. Twenty subjects were on d-drug treatment at enrollment. These subjects were treated for a median of 3.7 years (interquartile range 3.0–5.3 years). Seven of these subjects developed a DSP; 57% (4/7) developed a DSP within the first year of follow-up. A higher percentage of subjects with either a history of d-drug treatment but not on a d-drug at enrollment or currently on d-drug therapy developed a symptomatic DSP (9/35, 26%) compared to those who were never treated with a d-drug (2/26, 8%, Fig. 1). The difference, however, did not reach statistical significance (p = 0.10). The small number of outcomes prevented further analysis to look for trends based on years of exposure or type of d-drug.
Discussion
This study followed 61 HIV-positive subjects on HAART and without DSP at enrollment over 4 years for the development of DSP. Our study included subjects never treated with a d-drug and those who had tolerated d-drug treatment without developing a toxic DSP. Our finding suggests that incident DSP risk is not increased in subjects who currently or previously tolerated d-drugs without developing a toxic DSP.
Prior to HAART, HIV-associated DSP occurred in 35% of patients infected with HIV and was associated with advanced immunosuppression. 5 The association of high plasma HIV viral load and reduced CD4+ lymphocyte cell count with HIV-associated DSP was consistent with this observation. 7 In the HAART era, DSP continues to be a common neurologic complication of patients infected with HIV despite suppression of HIV viral loads and maintenance of higher CD4+ lymphocyte cell counts. 1 This continued prevalence of DSP may partially be secondary to residual HIV-induced effect on peripheral nerves perhaps mediated by proinflammatory cytokines (e.g., tumor necrosis factor-α, interleukin-1, interleukin-6) secreted by HIV-infected macrophages. 19
In the HAART era, particular antiviral therapies, collectively known as “d-drugs,” were found to cause a toxic-DSP, occurring in up to 34% of patients. 8 –11 Classically, symptoms are usually temporally related to the start of therapy occurring in the first 6 weeks of exposure. Although symptomatic improvement can occur with very early drug cessation, there is usually incomplete resolution of symptoms and signs. 19 It is currently believed that the toxic DSP related to d-drug therapy is the result of mitochondrial toxicity related to the inhibition of mitochondrial DNA polymerase-gamma. 11
It is well established that DSP continues to be a prevalent neurologic complication associated with HIV infection in the HAART era, occurring in 36–62% of subjects infected with HIV. 1 –4 Little is known about the incidence of DSP in the era of HAART. Lichtenstein et al. 13 reported an incidence of five cases per 100 person-years from the HIV Outpatient Study (HOPS). However, they included only symptomatic DSP in their study, and our study's higher incidence rate may have resulted from our inclusion of both asymptomatic and symptomatic DSP cases. Our cohort was also enriched with older individuals. Our results do not contradict the higher rates of incident DSP (36–54%) reported in cohort studies from sub-Saharan Africa 20,21 where d4T was used given that our sample included subjects who had already demonstrated an ability to tolerate these drugs without developing DSP.
In the HAART era, the risk factors associated with prevalent DSP in subjects infected with HIV have changed. High plasma HIV viral loads and decreased current CD4+ cell counts are no longer associated with DSP. 2,22 A prior study from HAHC has identified age greater than 50 years and nadir CD4+ cell counts below 200 cells/μl as a risk factor associated with prevalent DSP. 16 This has been confirmed in other studies. 13,23,24
Our study found that nadir CD4+ cell counts and age are also significant risk factors for incident DSP in the era of HAART (Tables 2 and 3). The relationship of nadir CD4+ cell counts with incident and prevalent cases of DSP is possibly the result of damage to the peripheral nervous system that occurred at a time when the plasma HIV viral load was higher and CD4+ cell counts were lower. 13
Since the introduction of HAART in 1996, individuals infected with HIV are living longer and understanding the interaction between age and medical/drug-related comorbidities will become increasingly important. Cherry et al. 25 propose that the development of DSP could be the result of reduced neurologic reserve. In the aging HIV population, the peripheral nervous system will be affected by the inflammatory cascade from dysfunctional macrophages 19 in addition to other medical comorbidities, such as alcohol abuse, diabetes, B12 deficiency, and concurrent infection with hepatitis C virus. Mitochondrial dysfunction also occurs with increased age. 26 There have been concerns that older HIV-infected individuals treated with mitochondrial toxic d-drugs might decrease neurologic reserve, unmasking preexisting subclinical peripheral nerve damage due to HIV. 27,28
Symptomatic DSP is more common in older (≥ 50 years of age) HIV-infected individuals. 16 There was no significant difference in proportion of symptomatic DSP in the younger (<40 years of age) and older groups in our study, likely due the small number of this outcome. Although symptomatic neuropathy was not the primary outcome of this study, there was a trend toward a higher incidence of symptomatic DSP in d-drug-experienced subjects (9/35, 26%) versus those never treated with a d-drug (Fig. 1). Given that d-drugs, especially d4T, are still used in resource-limited settings 4,29,30 and some believe that the relationship between d-drugs and the development of symptomatic DSP results from the unmasking of preexisting subclinical peripheral nerve damage by the toxic agents, future studies to determine risk factors involved with the development of symptomatic DSP and progression of an asymptomatic to symptomatic DSP are warranted. Cherry et al. 30 and Affandi et al. 29 found that d4T neuropathy risk increases with age and height and suggest that prioritizing older and taller subjects to alternative antiviral therapies would be one inexpensive strategy to reduce DSP in resource-limited settings.
Our study did not find a significance difference in incidence of DSP between subjects never treated with a d-drug and those with a history of d-drug treatment. This can in part be explained by a selection bias. Subjects selected for this analysis were free of DSP at enrollment, and those with a history of d-drug exposure who had already developed a DSP were excluded. Therefore, this population was likely relatively resistant to d-drug toxicity, which may explain our findings (Tables 2 and 3). This is consistent with the finding from Lichtenstein et al. 13 that if DSP does not develop within the first year of use of d-NRTI it is less likely to occur in subsequent years. A recent study by Hung et al. 31 also found that subjects tolerating an initial trial of a d-drug did not have an increased risk of progression of neuropathic signs or symptoms compared to subjects not on a d-drug. Our finding adds to the literature by emphasizing that there is no ongoing residual increase in DSP risk for subjects who received and tolerated d-drugs in the past and that there appears to be no significant added DSP risk if d-drugs are continued in subjects currently tolerating d-drug. The former is important information for patients in developed countries many of whom received d-drugs in the past. The later is important information for subjects in resource-limited countries in assessing the risk of continued d4T use. Although d-drugs have other toxicities such as lipoatrophy associated with their use, 32 this information will assist physicians with treatment decisions in resource-limited settings where d-drugs remain one of the few affordable treatment options for individuals infected with HIV.
We recognize the limitations of the current study. First, there was a small number of outcomes in our study, and a type-II error may account for our finding that the risk of developing a toxic DSP may not increase with d-drug exposure. The small number of outcomes also prevented subgroup analysis between asymptomatic and symptomatic DSP. Second, DSP was a clinical diagnosis, and the incidence of DSP may have been underestimated since electrophysiologic studies or skin biopsies were not performed. Third, since asymptomatic and symptomatic DSP were the outcomes of interest, the reliability of the examination could be a concern. In the HAHC, however, neurologic examinations were performed by the same two investigators (V.V., M.R.W.). One was a neurologist and the second an internist with semiannual quality assurance on the technique. Fourth, subjects were not systematically evaluated for B12 deficiency or hepatitis C virus status, and we were unable to determine the contribution of these confounders to the risk of incident DSP. Fifth, the reason for the discontinuation of the d-drug was not collected, and this could account for the decreased incidence associated with subjects who had a history of d-drug treatment but were not on a d-drug at enrollment.
In summary, our findings show that current or prior use of d-drugs is not a significant risk factor for incident DSP in subjects who had previously tolerated d-drug treatment without developing a toxic DSP. Age and nadir CD4+ count appear to be important risk factors for incident DSP. Perhaps future studies should address whether initiating antiviral therapy earlier or tailoring therapy based on age may minimize incident DSP risk.
Footnotes
Acknowledgments
This study was supported by research Grants R01NS063932 NINDS/NIH “HIV and Global Drug Therapies: Peripheral Neuropathy Complications and Mechanisms,” P200RR011091 NCRR/NIH Clinical Research Center, U54 NS4 NINDS/NIH NeuroAIDS Specialized Neuroscience Research Program, R25 RR019321 Clinical Research Education and Career Development (CRECD) in Minority Institutions “Master in Biomedical Science Program in Clinical Research,” K07 GM072884 Curriculum Development Award in Interdisciplinary Research “PhD in Biomedical Science Program in Clinical Research.” Thank you to Roseanne Harrigan, Ed.D. and the members of the Master and Ph.D. in Biomedical Science Program in Clinical Research for reviewing this chapter. Statistical analysis was performed by James Davis, Ph.D., John A. Burns School of Medicine, University of Hawaii, Honolulu, HI.
Author Disclosure Statement
Dr. Valcour is a consultant for Merck and Glaxo Smith Kline. Dr. Shikuma receives research/training funding support from Pfizer, Merck, and Gilead Pharmaceuticals, and has served on an advisory board for Glaxo Smith Kline.