
Abstract
Each cell in HIV-associated primary central nervous system lymphoma (PCNSL) harbors latent EBV. Notably, the triggering of TLR9, a key event in HIV pathogenesis, also promotes EBV latency and transformation. We hypothesized that because only a minority of HIV-infected patients develops PCNSL, their B cells exhibit aberrant signaling responses to TLR9 triggering. We found higher levels of IL-6, CD80, and CD86 expression at baseline in B cells of those patients than in B cells of matched controls, whereas TNF-α expression was lower. Notably, on TLR9 triggering with CpG 2006, CD80 and TNF-α were up-regulated to a lesser extent in B cells of the former than in those of matched controls. The reduced up-regulation of CD80 might be explained by its higher baseline expression resulting in a more blunted response rather than a specific deficit of the signaling response to TLR9 triggering. However, this cannot explain the blunted TNF-α response, which warrants further investigation. Finally, since increased IL-6 expression is linked to EBV-associated Hodgkin's lymphoma, the enhanced baseline expression of IL-6 might be important in the pathogenesis of PCNSL in HIV-infected patients.
P
Each cell of HIV-associated PCNSL harbors EBV transcripts. 6 The EBV gene product latent membrane protein 1 (LMP1) most likely contributes to malignant transformation by up-regulating expression of B cell lymphoma protein (BCL)-2. 7 EBV may also help B cells, which are rarely found in normal central nervous system (CNS) parenchyma, 8,9 to enter the CNS due to their EBV-associated activation. Furthermore, the absence of EBV-specific CD4+ T cells in AIDS patients with PCNSL 10 implicates EBV as a critical pathogenic factor in HIV-associated PCNSL.
Sustained immune activation is a major factor in the progressive immunodeficiency in HIV infection. Activation may result from an increased influx of microbial components leaking from the gut, 11 the HIV Nef protein, 12 or opportunistic infections. 13 The influx of microbial components and opportunistic infections also contributes to immune activation by triggering Toll-like receptors (TLRs), such as TLR 7/8. 14 TLRs belong to the family of pattern recognition receptors that recognizes conserved motifs of microorganisms. 15 Triggering TLRs leads to complex signaling cascades, which result in cellular activation, secretion of cytokines, and up-regulation of MHC class molecules. The significance of TLR triggering in HIV infection is underscored by the effects of TLR8 and 9 polymorphisms on its outcome. 16,17
TLR signaling and immune activation are also linked to EBV-associated lymphomagenesis. 18 Endemic Burkitt's lymphoma occurs in areas in which malaria is holoendemic. A glycosylphosphatidylinositol anchor on the cell surface of the malaria-causing protozoon, Plasmodium falciparum, is recognized by TLR2 and TLR4. 19 Furthermore, the plasmodial DNA itself exhibits a strong activation potential by triggering TLR9. 20 Overall, immune activation is thought to contribute to the development of Burkitt's lymphoma by stimulating the proliferation and expansion of the pool of EBV latently infected memory B cells, thereby increasing the risk of emergence of a malignant B cell clone. 18 TLR9 triggering on EBV-infected memory B cells may also result in their terminal differentiation into plasma cells and lytic EBV infection. Release of infectious EBV virions may, in turn, infect more B cells and increase the pool of latently infected B cells. We recently showed that triggering of TLR9 suppresses lytic EBV replication in cord blood cells infected ex vivo with EBV and in anti-IgG-stimulated Akata Burkitt's lymphoma cells harboring latent EBV. 21 In this way, TLR9-dependent immune activation may contribute to EBV-associated lymphomagenesis by suppressing lytic infection and promoting latent EBV infection with a uniquely high transformation potential. This phenomenon may be even more pronounced in advanced HIV infection when EBV-specific immune responses are lost. Although only a few HIV-infected patients develop PCNSL, TLR9 agonists may favor EBV latency and the development of EBV-associated PCNSL.
We hypothesized that B cells from HIV-infected patients with PCNSL respond more to TLR9 agonists than B cells from patients without PCNSL. To test our hypothesis, we examined the responsiveness of B cells obtained from HIV-infected patients with PCNSL, matched HIV-infected patients without PCNSL, and healthy volunteers to a TLR9 agonist by immunostaining and flow cytometry. Selection of patients from the Swiss HIV Cohort Study (SHCS) was based on documentation of PCNSL and the availability of cryopreserved peripheral blood mononuclear cells (PBMCs) with a laboratory date (i.e., date of blood withdrawal) prior and as close as possible to the date of PCNSL diagnosis. For simplicity reasons, we use the term “patients with PCNSL” for this patient group. Matching criteria for the HIV-infected controls (twice as many as patients with PCNSL) were the following: (1) gender, (2) age at the date of blood sample collection (±3 years), (3) date of blood sample collection (±1 year), (4) CD4+ T cell counts (±50 cells/μl), and (5) plasma HIV RNA copy numbers. The demographics of the HIV-infected patients with PCNSL and matched HIV-infected controls are shown in Table 1. The profiles of CD4 count, antiretroviral therapy (ART), and viral load (if known) over time are shown in Supplemental Fig. 1 (see
NA, not available.
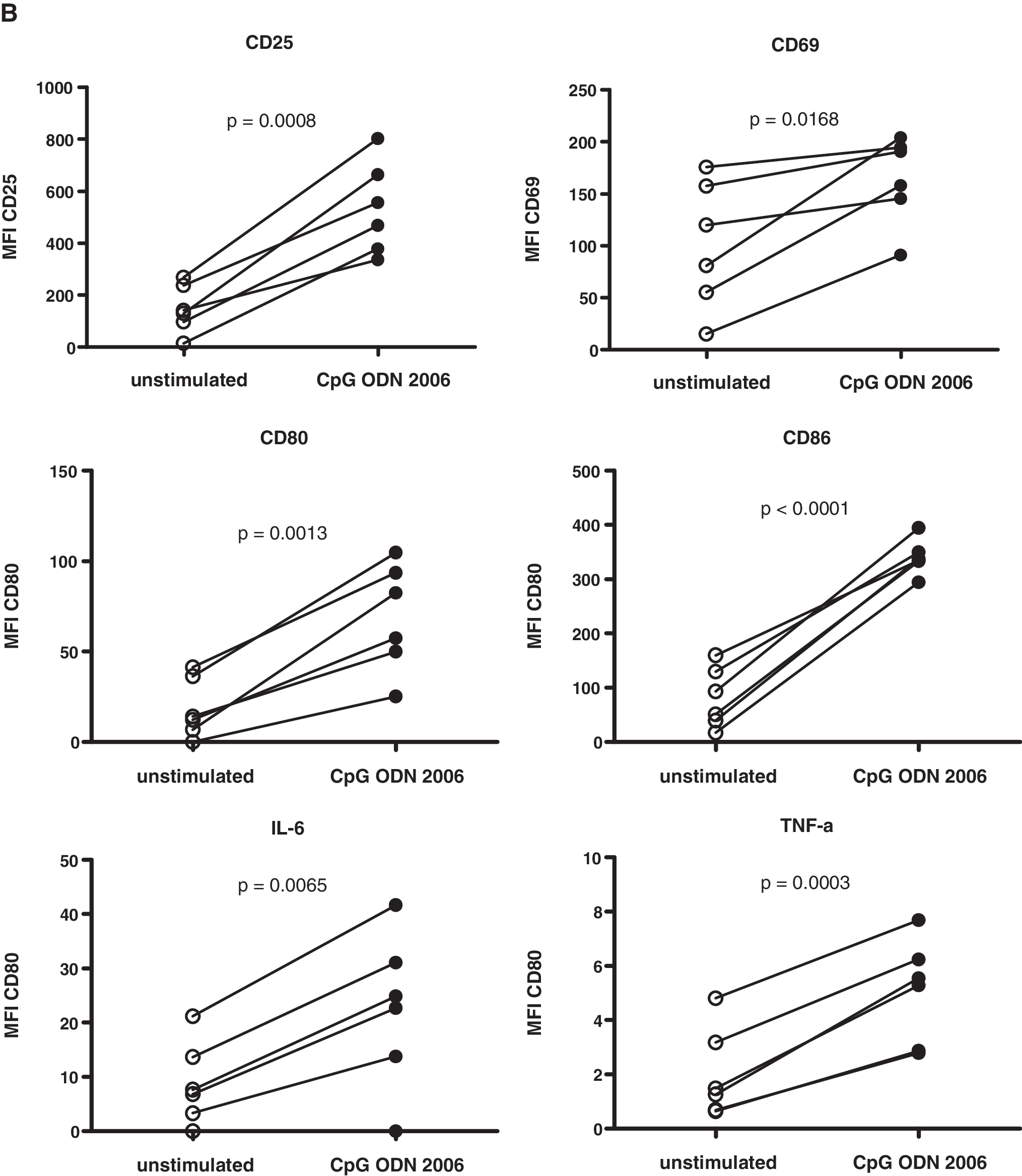
We validated our assays with cryopreserved PBMCs from HIV-negative volunteers. We observed an increase in the percentage of CD19+ cells expressing CD25, CD69, CD80, CD86, interleukin (IL)-6, and tumor necrosis factor (TNF)-α (Fig. 1A) and in the mean fluorescence intensity (MFI; Fig. 1B) in response to the TLR9 agonist CpG 2006, confirming our approach. 22 These markers were used previously to measure the activity of TLR9 agonists. 23,24
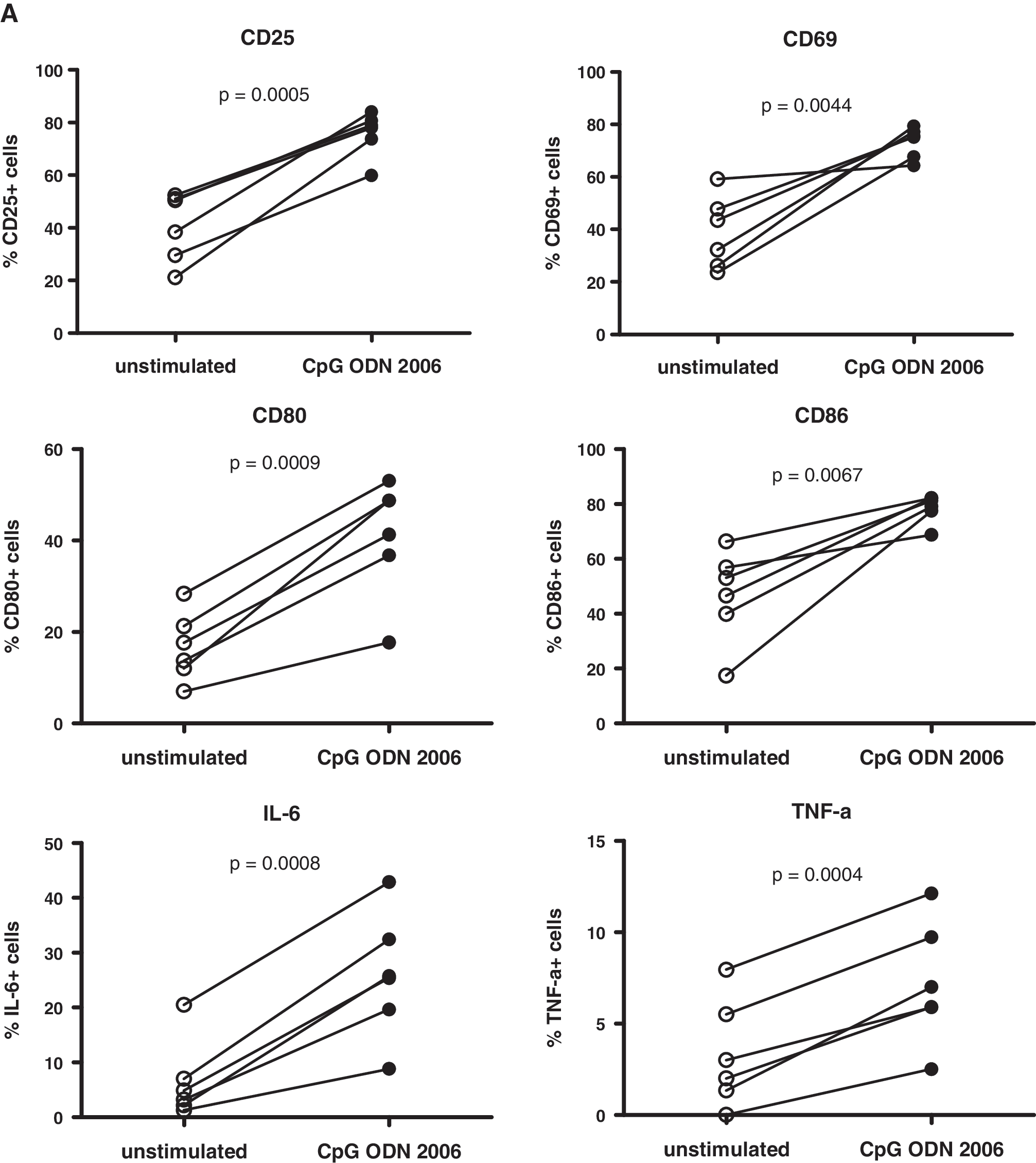
Up-regulation of activation and costimulatory markers and of proinflammatory cytokines on/in B cells after stimulation of PBMCs from HIV-negative donors with the TLR9 agonist CpG 2006. Cryopreserved PBMCs were rapidly thawed and recovered in RPMI medium containing 10% FCS and 10 U/ml IL-2 (= culture medium), centrifuged, resuspended in culture medium at 2.5 × 106 cells/ml, and put into 96-well U-bottom plates at 5 × 105 cells/well. For analysis of activation and costimulatory markers, cells were stimulated with CpG 2006 (Microsynth GmbH, Balgach, Switzerland) at 3 (μg/ml for 24 h; control cells were not stimulated. For analysis of cytokines, cells were cultured for 18 h and then stimulated with CpG 2006 at 3 (μg/ml for another 6 h in the presence of brefeldin A (GolgiPlug; BD Pharmingen); control cells were not stimulated, but treated with brefeldin A. Cells were then analyzed for B cell surface marker/cytokine expression by flow cytometry with antihuman CD19 (conjugate: FITC), CD25 (PE), CD69 (PE-Cy5), CD80 (PE), CD86 (PE-Cy5), IL-6 (PE), and TNF-α (PE-Cy7) monoclonal antibodies (mAbs; all Abs from BD Pharmingen) on a FACSCalibur (BD Biosciences) or CyAn ADP (Beckman Coulter) flow cytometer. Data were analyzed with FlowJo software (Tree Star). Percentage (
Because of the limited number of available cells, we used stimulation of PBMCs instead of purified B cells. To validate the use of PBMCs we isolated B cells from an HIV-negative healthy donor and compared the up-regulation of the above-mentioned markers/cytokines on/in B cells after stimulation with CpG 2006 between unfractionated PBMCs and isolated B cells. We found that up-regulation of marker/cytokine expression was similar between PBMCs and B cells (Supplemental Fig. 2; see
In HIV-infected individuals, the composition of the B cell compartment is altered with an expansion or contraction of one or several of the various B cell subpopulations. 28 In the present study, we were not able to differentiate between naive and memory B cells due to the limited number of available cells. However, all patients in this study showed a very advanced stage of HIV disease (Table 1 and Supplemental Fig. 1). We have no reason to assume that the two groups investigated differ in their B cell composition, and, therefore, this is unlikely to have an impact on our data.
The baseline expression levels of most of the investigated markers/cytokines were similar in HIV-infected patients and healthy volunteers. However, CD86 was expressed at higher levels in HIV-infected patients without PCNSL, and the MFI was higher in HIV-infected patients with PCNSL (data not shown). Binding of the costimulatory markers CD80/CD86 to CD28 results in activation of CD4+ T cells to CTLA-4 and leads to their inhibition. 29 CTLA-4 has a much higher affinity for CD80/CD86 than CD28. Because CTLA-4 is up-regulated on HIV-specific CD4+ T cells in progressive disease, 30 the increased expression of CD80/86 on B cells by binding to CTLA-4 on CD4+ T cells may contribute to the overall immune dysfunction in HIV disease. Similar increases in CD86 expression in B cells from HIV viremic patients were previously reported. 31
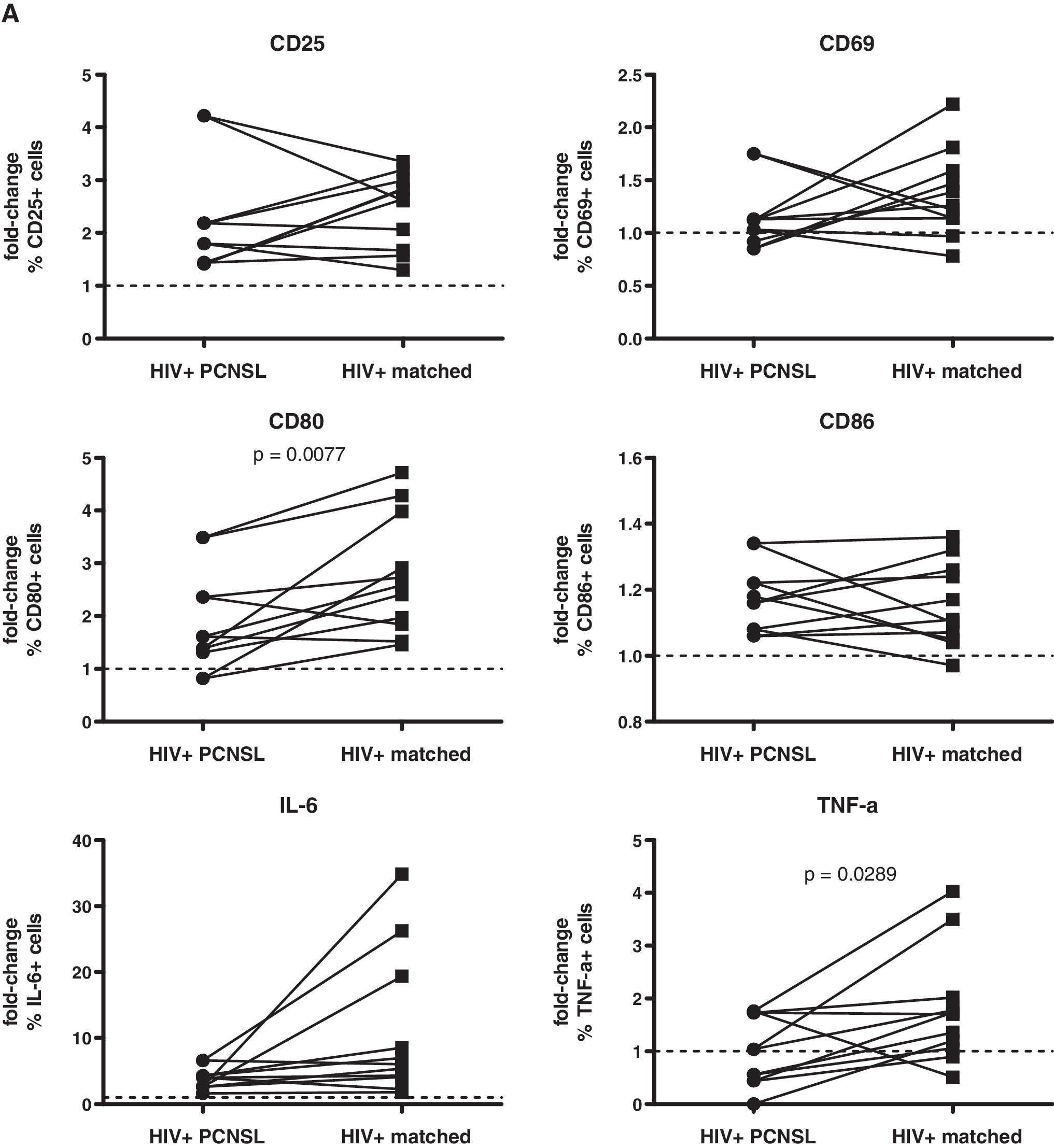
We then compared the fold-increase of marker/cytokine expression on/in B cells after stimulation with CpG 2006, initially without considering the pairwise matching of HIV-infected patients (for statistical analysis see the figure legends). B cells from HIV-infected patients reacted with a similar up-regulation of CD25 and intracellular accumulation of IL-6 as those from healthy volunteers. In contrast, fold-increases of B cell expression were lower for CD69, CD80, CD86, and TNF-α in HIV-infected patients than in healthy volunteers (Fig. 2). Notably, up-regulation of CD86 is lower in HIV-infected patients. 32 The blunted up-regulation of CD86 may be explained by its higher baseline expression level, which may limit the magnitude of absolute increase. Impaired B cell reactivity is well documented in HIV infection when assessing vaccine responses. 33 –35 Notably, in patients with PCNSL the fold-increases were even less pronounced for CD69 and TNF-α than in patients without PCNSL (see below). Similar to our data, B cell receptor/CD40 stimulation also results in a reduced up-regulation of CD25, CD80, and CD86 in HIV viremic patients as compared to HIV-negative volunteers. 31 Thus, B cells in HIV infection appear to have a generally reduced reactivity, independent of the stimuli.
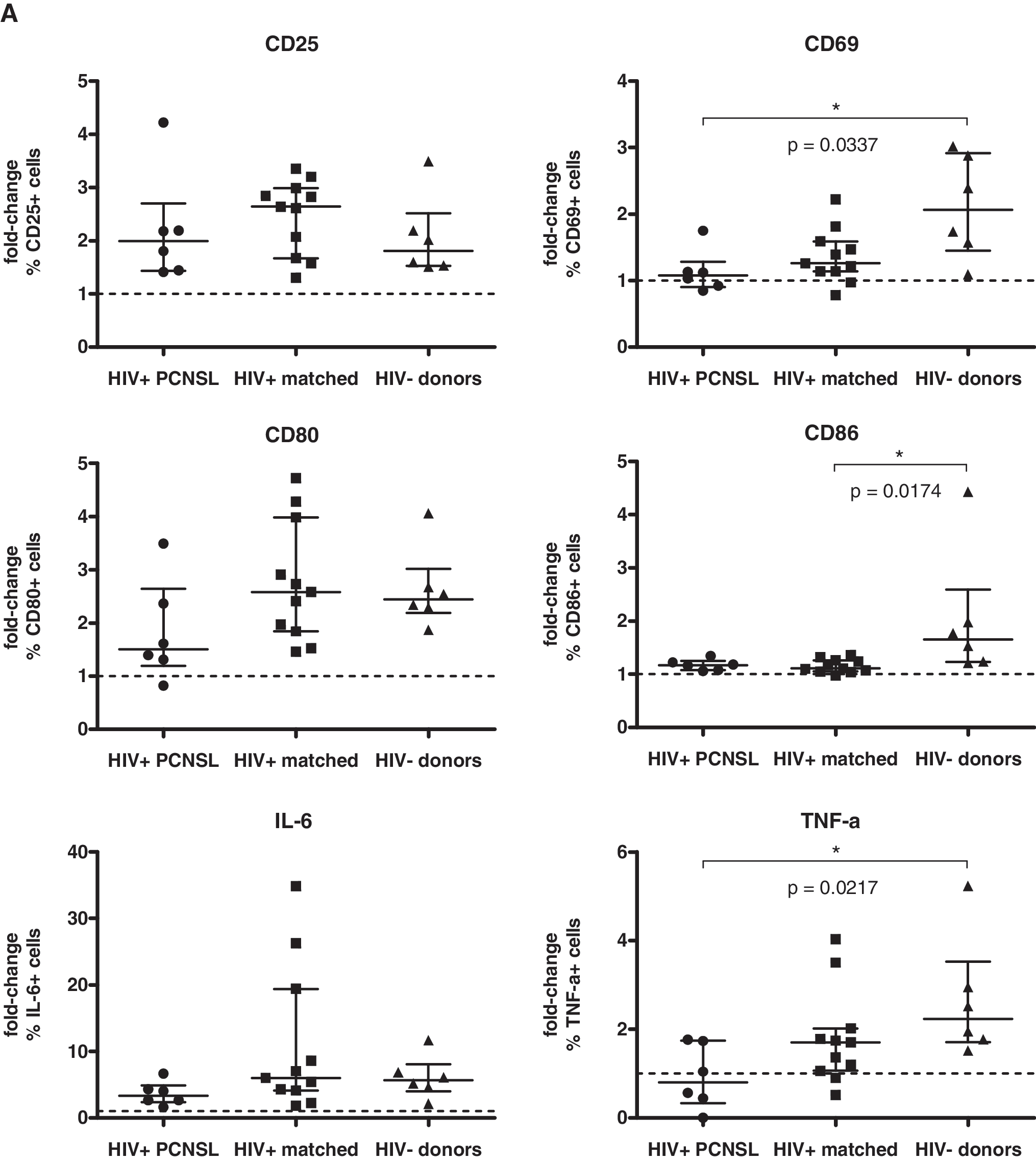
Fold-increase of marker/cytokine expression in B cells from HIV-infected patients with PCNSL and those without PCNSL compared to that of HIV-negative donors after stimulation of PBMCs with CpG 2006. PBMCs were stimulated and analyzed as described in Fig. 1. Data are given as fold-change of percent expression (

The key interest of our work was to compare the reactivity to TLR9 triggering of B cells from HIV-infected patients with PCNSL to those from HIV-infected patients without PCNSL. We thus reanalyzed the data, now using paired Student's t test, considering the pairwise matching. At baseline, we observed significantly higher levels of IL-6 expression (in percentage of cells and MFI) and CD86 MFI and significantly lower levels of TNF-α expression (in percentage of cells and MFI) by B cells from the HIV-infected patients with PCNSL as compared to the matched controls (Fig. 3). CD80 expression (MFI) at baseline was also increased, but this difference did not reach statistical significance.
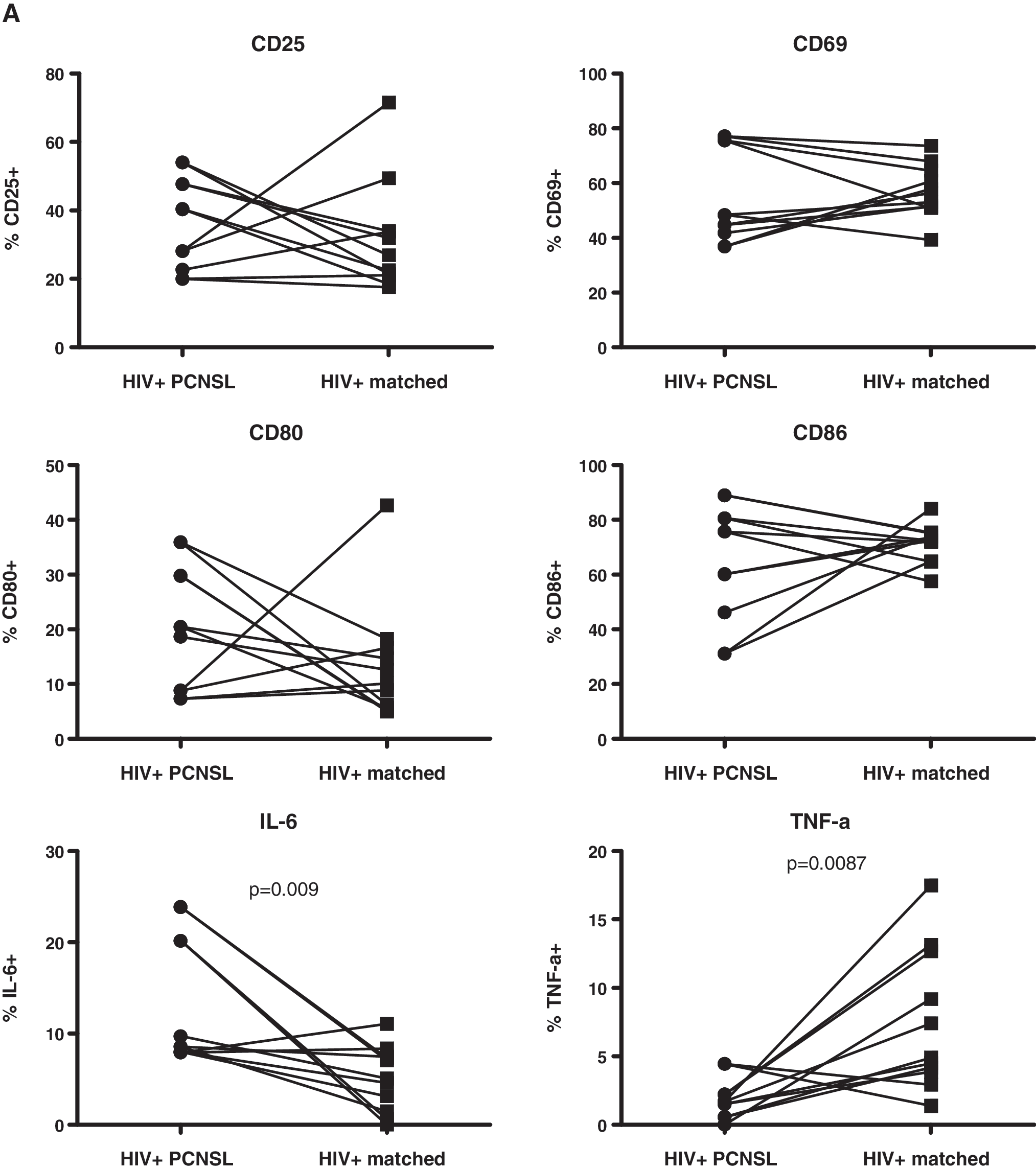
Comparison of baseline expression of B cell markers between HIV-infected patients with PCNSL and matched controls. PBMCs were cultured without stimulation and analyzed as described in Fig. 1. Percentage (
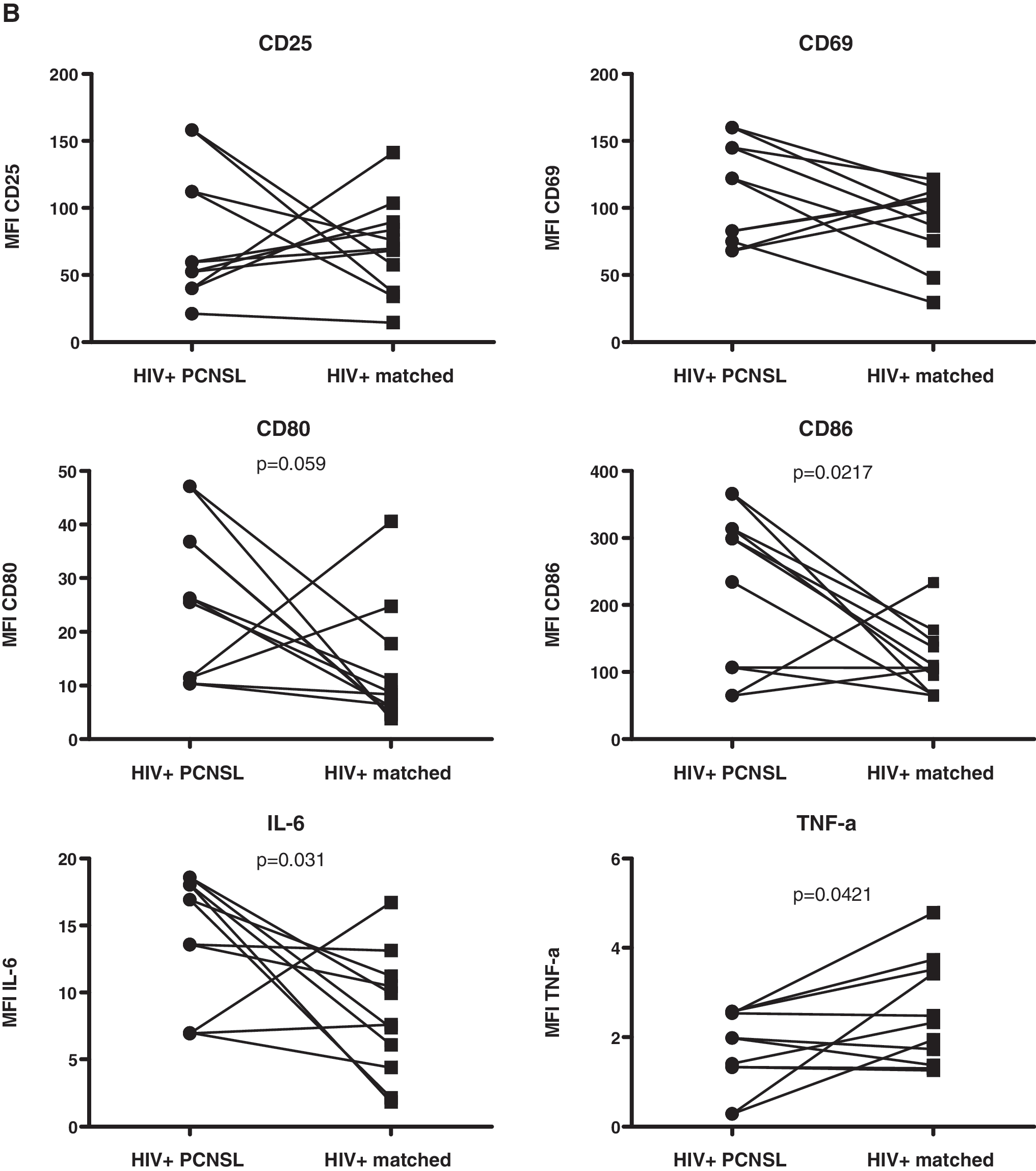
As noted above, the increased expression of CD80/CD86 may result in an increased binding of B to T cells via the interaction of these markers with CTLA-4, contributing to a decreased T cell reactivity and potentially to a decreased EBV-specific T cell response. The obvious lack of EBV-specific CD4+ T cells 10 together with the increased expression of CD80/CD86 may represent a unique risk profile for those HIV-infected patients with PCNSL. Studies performed in the 1990s showed that elevated serum/plasma IL-6 levels preceded the diagnosis of AIDS-related lymphoma. 36,37 Furthermore, the higher IL-6 expression levels at baseline is similar to a report linking increased IL-6 levels to the risk for Hodgkin's lymphoma, 38 another EBV-associated neoplasia. Thus, IL-6 may be implicated in the etiology of PCNSL. TNF-α induces IL-6 39 ; thus, increases of IL-6 should parallel increases in TNF-α. However, this was not the case: patients with PCNSL showed reduced expression of TNF-α. The significance of this observation is unknown. We cannot exclude the possibility that there is an increased production of TNF-α in other, non-B cell subsets such as T cells, and that this may compensate for the lower TNF-α production in B cells.
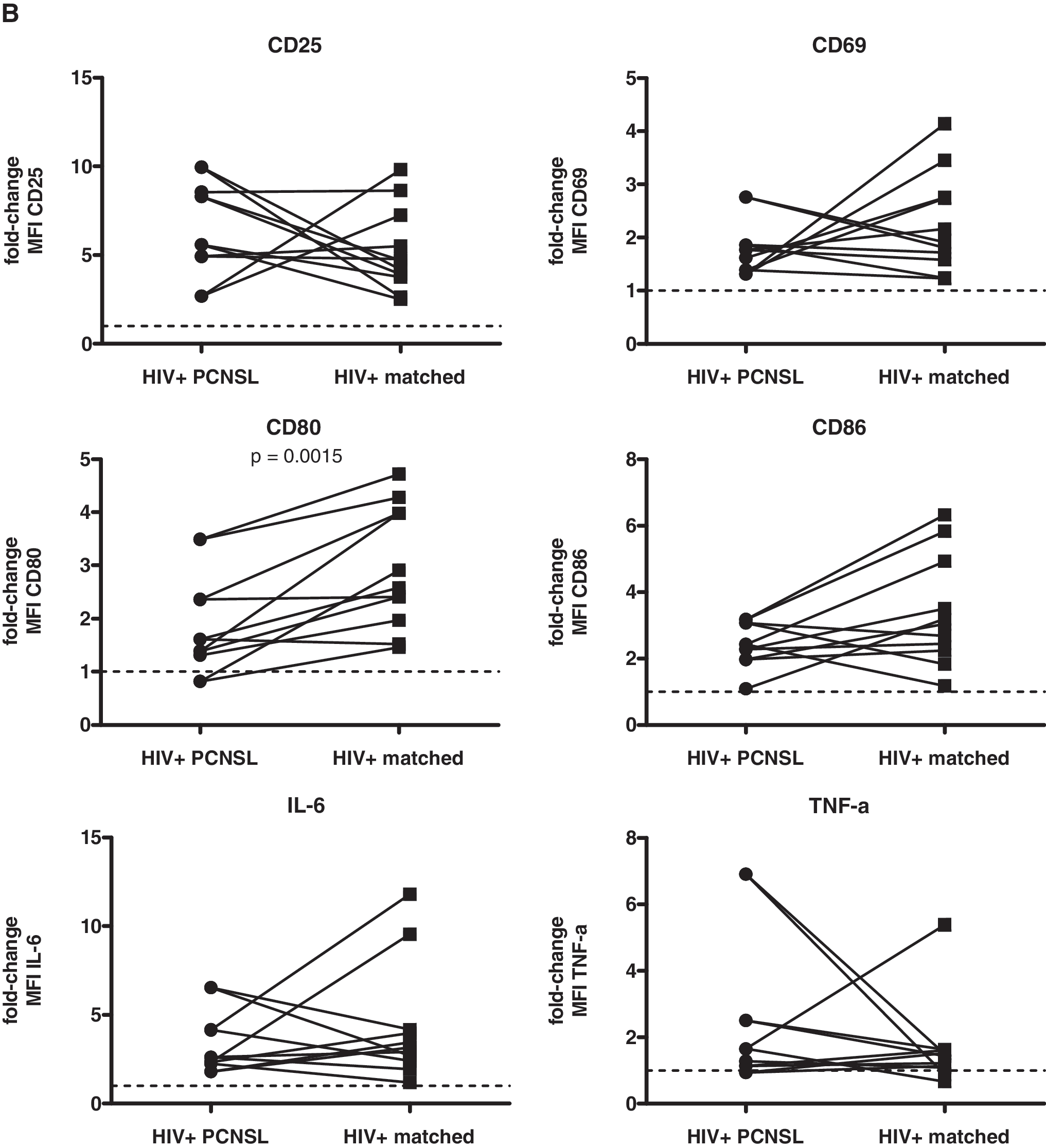
After treatment with CpG 2006, we observed that B cells from patients with PCNSL showed less pronounced up-regulation of CD80 expression and TNF-α production than B cells from matched controls (Fig. 4). Because there was a tendency toward increased baseline levels of CD80 in HIV-infected patients with PCNSL, the lack of its further increase subsequent to stimulation with CpG 2006 speaks in favor of a threshold of maximal expression. This cannot explain the reduced TNF-α response since its production was decreased at baseline in B cells of patients with PCNSL compared to the matched controls, suggesting a specific disturbance resulting in impaired TNF-α production in patients with PCNSL.
Comparison of responsiveness of B cells to TLR9 stimulation between HIV-infected patients with PCNSL and matched controls. PBMCs were stimulated and analyzed as described in Fig. 1. Data are given as fold-change of percent expression (
In conclusion, we found that B cells from HIV-infected patients with PCNSL display an activated phenotype (increased IL-6 and CD86 expression at baseline) and have a blunted TNF-α response to TLR9 triggering. The enhanced baseline expression of IL-6 might be important in the pathogenesis of PCNSL in HIV-infected patients since increased IL-6 expression is linked to EBV-associated Hodgkin's lymphoma. The impaired TNF-α response despite a decreased baseline expression is intriguing and warrants further investigation. A study with a larger sample size of HIV-infected patients with PCNSL is not realistic with most HIV-infected patients being under ART and because of the very low incidence of PCNSL. However, it would be of great interest to examine whether B cells of patients with other forms of EBV-associated lymphoma and in particular with endemic Burkitt's lymphoma have aberrant signaling responses to TLR9 triggering.
Footnotes
Acknowledgments
This study has been financed in the framework of the Swiss HIV Cohort Study, supported by the Swiss National Science Foundation and the Julius Müller Stiftung. The members of the Swiss HIV Cohort Study are M. Battegay, E. Bernasconi, J. Böni, H.C. Bucher, P. Bürgisser, A. Calmy, S. Cattacin, M. Cavassini, R. Dubs, M. Egger, L. Elzi, M. Fischer, M. Flepp, A. Fontana, P. Francioli (President of the SHCS), H. Furrer (Chairman of the Clinical and Laboratory Committee), C.A. Fux, M. Gorgievski, H.F. Günthard (Chairman of the Scientific Board), H.H. Hirsch, B. Hirschel, I. Hösli, C. Kahlert, L. Kaiser, U. Karrer, C. Kind, T. Klimkait, B. Ledergerber, G. Martinetti, N. Müller, D. Nadal, F. Paccaud, G. Pantaleo, A. Rauch, S. Regenass, M. Rickenbach (Head of the Data Center), C. Rudin (Chairman of the Mother & Child Substudy), P. Schmid, D. Schultze, F. Schöni-Affolter, J. Schüpbach, R. Speck, B.M. de Tejada, P. Taffé, A. Telenti, A. Trkola, P. Vernazza, R. Weber, and S. Yerly.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.