
Abstract
The next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) etravirine (TMC125) has demonstrated durable virologic efficacy in clinical trials involving >1000 treatment-experienced, NNRTI-resistant, HIV-1-infected patients. In this clinical safety review, we show etravirine to be well tolerated with a proven safety record. The nature and magnitude of adverse events observed during treatment suggest that etravirine may offer improved tolerability over existing antiretrovirals, including NNRTIs. Notably, adverse events reported with etravirine treatment are generally mild to moderate in severity. Rash has been shown to occur with a higher incidence in etravirine-treated patients versus placebo, but cases are generally mild to moderate, occur within the first few weeks, and resolve with continued use. In addition, the rate of adverse event-related discontinuations is low with etravirine. In summary, the safety and tolerability profile of etravirine, combined with its virologic efficacy, suggest that the drug may be a valuable option for treatment-experienced patients with HIV-1 infection.
Introduction
H
Nonnucleoside reverse transcriptase inhibitors (NNRTIs) have played a vital role in antiretroviral therapy for more than a decade because of their favorable tolerability profile, high potency, and convenient dosing. However, a single amino acid substitution in the HIV-1 reverse transcriptase enzyme can confer within-class cross-resistance and result in a significant decrease in HIV susceptibility to the first-generation NNRTIs nevirapine, efavirenz, and delavirdine. 1 –4 For these reasons first-generation NNRTIs are used primarily in treatment-naive patients.
The development of etravirine (ETR; TMC125) has extended the use of the NNRTI class to treatment-experienced adult patients with HIV that is resistant to first-generation NNRTIs. ETR exhibits potent and broad in vitro activity against HIV-1, including viral strains with NNRTI resistance-associated mutations. 5,6 It has demonstrated rapid and durable antiretroviral suppression in treatment-experienced patients. 7 –10 Forty-eight week data from the randomized, placebo-controlled Phase III DUET trials revealed that a significantly greater percentage of patients had their viral load reduced to less than <50 HIV-1 RNA copies/ml when treated with ETR plus a background regimen (composed of at least two NRTIs, ritonavir-boosted darunavir, and optional enfuvirtide) than when treated with placebo plus the background regimen (61% vs. 40%; p < 0.0001 vs. placebo). 11 The durability of the virologic response has since been confirmed by 96-week data from the Phase III DUET trials. After 96 weeks, 57% of ETR-treated patients had their viral load reduced to less than <50 HIV-1 RNA copies/ml compared with 36% of placebo-treated patients (p < 0.0001). 12
The aim of this review is to consider available clinical safety data for ETR compared with placebo. Where comparison with other antiretrovirals is required, historical control data from prescribing information and/or key clinical trials will be discussed.
Materials and Methods
Search strategy and selection criteria
Articles reporting the safety and tolerability of ETR were identified by searching MEDLINE (up to January 2009) and relevant HIV/AIDS congresses (over the period January 1996–January 2009) using key terms including but not limited to NNRTIs, TMC125, etravirine safety, tolerability, toxicity, rash, neuropsychiatric, lipid, and hepatic toxicity. All relevant English language articles identified were reviewed and pertinent information is included in this publication.
General safety and tolerability of ETR
In all clinical studies conducted to date involving over 1000 patients, ETR (at a dose of 200 mg bid in the DUET and TRIO trials, and dose range of 400–1200 mg bid for an earlier formulation in Phase IIb studies) has been generally well tolerated with good cross-study consistency in the incidence and types of adverse events seen. 7 –15 Adverse events reported with ETR in the pooled DUET trials were primarily mild to moderate in severity (grade 1 or 2) and occurred at a similar rate to placebo, with the exception of rash (Table 1).
AE, adverse event; BR, background regimen comprising darunavir/ritonavir and investigator-selected NRTIs. Enfuvirtide use was optional; ISR, injection site reaction; N = number of patients, n = number of patients with observations; SAE, serious adverse event.
Group term: combining all rash-related events.
In Phase IIb and III trials, the most commonly reported grade 1 or 2 adverse events occurring in ≥5% of ETR-treated patients included diarrhea, rash, nausea, headache, fatigue, injection-site reaction (associated with enfuvirtide use), and abdominal pain. 7 –11,13,14 With the exception of rash, all of these events occurred with a similar frequency to study placebo (administered in combination with an optimized background antiretroviral regimen). In the pooled 48-week DUET analysis, the frequency of grade 3 or 4 adverse events and serious adverse events was similar in patients receiving ETR or placebo: 33.2% vs. 34.9% of patients, respectively, for grade 3 or 4 adverse events and 19.7% vs. 23.3% of patients for serious adverse events (Table 1). 11 Of the serious adverse events observed in DUET, most were commonly associated with HIV-1 disease progression. No consistent pattern of type, frequency, or relation to ETR was noted. 7 –11,13,14 Adverse events led to trial discontinuation in a small proportion of patients receiving ETR in the Phase II and Phase III studies. For Phase IIb studies, the rate of adverse event-related discontinuation was in the range of 12.1–17.0%, 9,10 and for the pooled DUET studies this figure was 7.2%. 11 Significantly, the incidence of adverse events leading to discontinuation in patients treated with ETR in the DUET studies was similar to that in the placebo group (5.6%). Looking across the Phase IIb and Phase III trials, 1.7–4.4% of ETR-treated patients discontinued because of rash and 1.4–5.2% discontinued because of gastrointestinal events. 9 –11,13,14 In the 24-week analysis of the Phase II single-arm TRIO trial, one patient discontinued treatment, which included ETR, ritonavir-boosted darunavir, and raltegravir, due to grade 4 rash and fever. 15 Although the grade 4 rash was considered possibly related to treatment, it could not be attributed to a specific study drug.
All deaths reported in the ETR groups in the DUET trials at 48 weeks [12 patients (2.0%) vs. 20 patients (3.3%) in the placebo group] were considered by the investigators to be unrelated to ETR use (Table 1). 11
NNRTI-Associated AEs of Interest
Cutaneous hypersensitivity/rash
Treatment with first-generation NNRTIs can result in the development of adverse skin reactions, 16,17 with new-onset skin rash being observed in approximately 25% of patients. 18,19 For most patients, skin reactions are mild to moderate in severity, taking the form of a rash reaction, which resolves with continued treatment. 20,21 Women appear to be particularly susceptible to developing skin rash when receiving first-generation NNRTIs, such as nevirapine. 18,22 On rare occasions, severe skin reactions including Stevens–Johnson syndrome (0.3%) and toxic epidermal necrolysis have been seen with nevirapine 18 and to a lesser extent with efavirenz (0.1%). 19 These reactions have occasionally been fatal. 23 Interruption or discontinuation of nevirapine therapy is often caused by adverse skin reactions and has been reported to account for more than a third of therapy discontinuations. 24 Readministration of nevirapine to patients who have permanently discontinued due to rash is not recommended. 18
Although the majority of adverse events observed with ETR occur with a similar frequency to placebo, rash has been found to occur with a higher incidence in ETR treatment groups than in placebo groups. 7 –14 However, skin events occurring in patients receiving ETR are generally mild to moderate in severity and normally resolve with continued treatment. There was no evidence of a relationship between the dose of ETR and the incidence of rash or skin reactions in the Phase IIb studies. 9,10 With broader use of ETR following marketing authorization, severe cutaneous and hypersensitivity reactions, including Stevens–Johnson syndrome and toxic epidermal necrolysis, have been reported, although the incidence is rare. Since these reactions can be life-threatening, clinical guidance requires immediate discontinuation of the drug when severe reactions are suspected.
In the Phase III DUET trials, 48-week pooled data showed an overall incidence of rash of 19.2% in the ETR group versus 10.9% in the placebo group (p < 0.0001; Table 1). 11 It is worth noting that the incidence rate for rash in the ETR group in DUET was similar to that observed in the Phase II studies (∼20%). Rash was mainly reported as mild to moderate in severity (grade 1 or 2), with 1.3% of patients in the ETR group reporting a grade 3 rash; grade 4 rash was not reported in the ETR group. The number of rash-related serious adverse events was low in the ETR group [1 patient (0.2%) with grade 2 rash]. No cases of Stevens–Johnson syndrome, erythema multiforme, or toxic epidermal necrolysis were reported in the ETR group. One patient in the placebo group experienced grade 4 rash (Stevens–Johnson syndrome), which was considered likely related to an allergic reaction to trimethoprim/sulfamethoxazole. 25
Further analysis of 48-week pooled DUET data revealed that rash was generally characterized as maculopapular (no mucosal involvement, no blisters), occurred within the first 2 weeks of treatment (median onset day 14), and resolved with continued treatment (median duration 15 days) (Fig. 1). 25 Discontinuation of ETR therapy because of rash was infrequent, occurring in 2.2% of patients. Treatment discontinuation was protocol mandated for grade 3 or 4 rash. In the ETR treatment group, the incidence of rash was higher in women than in men (30.0% vs. 18.0%, p = 0.0365). 11 This gender difference was mainly observed in DUET-1 and was not observed in the placebo groups. However, no gender difference was observed in severity or frequency of treatment discontinuation due to rash. The median duration of rash in the ETR group was 14.5 days for women and 15 days for men.

Incidence of rash in the Phase III DUET trials (pooled 48-week analysis). BR, background regimen.
No relationship was observed between the incidence of rash and baseline CD4 cell count (similar to findings in the TMC125-C203 and TMC125-C223 studies 9,10 ) or ETR pharmacokinetic exposure. 25 Furthermore, patients with a history of NNRTI-related rash were not predisposed to developing rash with ETR. In summary, rash did occur more frequently with ETR than with placebo in the DUET trials but was mild to moderate in severity, occurred early, and infrequently led to treatment discontinuation.
Nervous system and psychiatric adverse events
The most commonly reported adverse events associated with efavirenz are neuropsychiatric events, which have been reported to affect 40.0–70.0% of patients. 20,26,27 Efavirenz prescribing information reports that following treatment, 52.7% of patients present with nervous system events compared with 24.6% of patients receiving control regimens. 19
Available data suggest that ETR is not associated with neuropsychiatric events. In clinical trials, all nervous system and psychiatric events were recorded as spontaneous adverse events; no questionnaires were used. In the Phase IIb TMC125-C203 study that assessed the safety and tolerability of ETR compared with placebo, neuropsychiatric adverse events generally occurred with an incidence similar to placebo. 9 The most commonly reported nervous system disorders (occurring in >2% patients) during ETR treatment were headache and dizziness. These events occurred primarily within the first 2–4 weeks of treatment. The most commonly reported (≥2% patients) psychiatric adverse events included insomnia, depression, anxiety, and sleep disorder. 9 When observed, neuropsychiatric events were generally mild to moderate in severity (grade 1 or 2), with grade 3 or 4, or serious adverse events accounting for only 2% of adverse events in the ETR group. Grade 2–3 nervous system disorders considered at least possibly related to ETR treatment occurred in 5% of patients (n = 9; three patients had grade 2 headache, two had grade 2 dizziness, one each had grade 3 dizziness, grade 2 dysgeusia, and grade 3 tension headache, and another patient had grade 2 headache, grade 2 dizziness, and a grade 2 tremor). Grade 2 psychiatric disorders considered at least possibly related to ETR treatment occurred in <2% of patients (n = 3), and included mood swings (n = 1), abnormal dreams (n = 1), and a confusional state (n = 1). No grade 4 treatment-related nervous system disorder or grade 3–4 treatment-related psychiatric disorder was reported. Neuropsychiatric adverse events resulted in treatment discontinuation in only 3% of patients; most events resolved without intervention.
Pooled 48-week safety data from the DUET-1 and −2 trials confirm these Phase IIb findings and show that the incidence of neuropsychiatric adverse events did not differ significantly in patients receiving ETR treatment compared with patients in the placebo group (Fig. 2). 11 Nervous system events were generally mild to moderate in severity and infrequently led to discontinuation. Of 599 ETR-treated patients, 17.2% reported nervous system events compared with 19.7% of patients receiving placebo (p = 0.266). Similarly, psychiatric events were equally balanced across treatment groups, with 16.7% of ETR patients exhibiting psychiatric events compared with 19.5% of patients in the placebo group (p = 0.204).
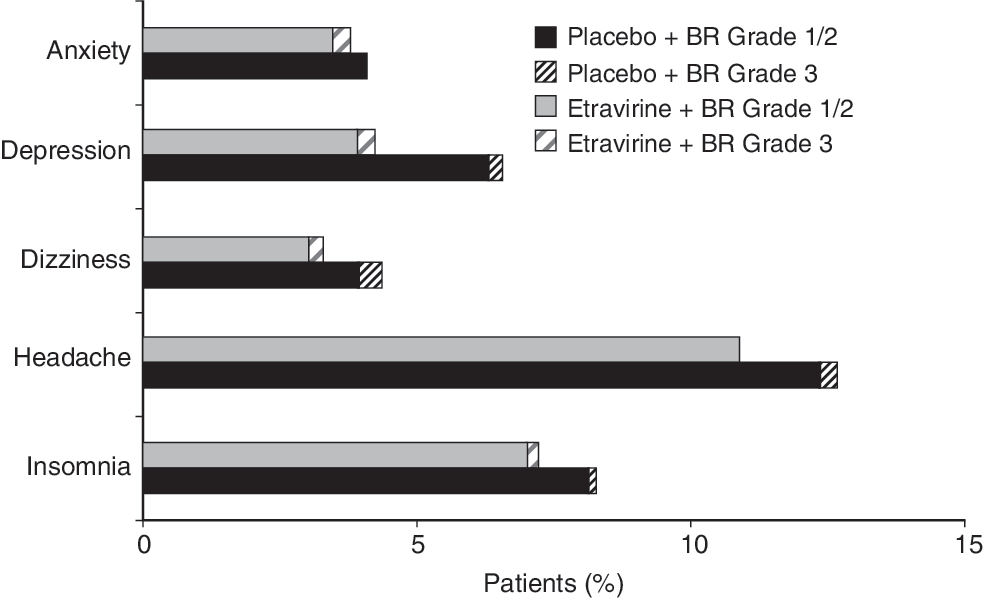
Incidence of the most common (occurring in ≥1% of patients in the etravirine group) neuropsychiatric adverse events of interest in the Phase III DUET trials (pooled 48-week analysis). BR, background regimen. Note: One patient in the placebo group had a grade 4 neuropsychiatric adverse event.
In addition, the psychiatric disorders of greatest frequency (reported in ≥1.0% of ETR patients) were insomnia (7.2% vs. 8.3% in the ETR vs. placebo groups, respectively), depression (4.2% vs. 6.6%), anxiety (3.8% vs. 4.1%), and sleep disorder (1.3% vs. 0.8%). 28 Nightmares occurred in 0.3% and 0.2% of ETR and placebo patients, respectively, and abnormal dreams in 0.5% and 0.8% of patients (Fig. 2). There were no episodes of hallucinations, suicidal ideation, or manic symptoms in the ETR group. In the pooled trials, the central nervous system (CNS)-related adverse events observed were mostly grade 1 or 2, with only 0.5% of ETR-treated patients reporting grade 3 neuropsychiatric disorders (vs. 2.0% of placebo-treated patients). No patients receiving ETR experienced grade 4 CNS-related events. No treatment discontinuations were associated with nervous system adverse events in the ETR group and only one patient (0.2%) in the ETR group discontinued as a result of psychiatric adverse events. 28
The frequency of neuropsychiatric events in DUET was higher in patients with a history of psychiatric disorders; however, incidences in this subgroup of patients were similar in the ETR and placebo treatment groups. 28
Hepatic toxicity
Another important consideration when selecting a new antiretroviral regimen is the potential for hepatic toxicity. 17,29 Liver toxicity has been associated with nevirapine and efavirenz, 18,19,30 with a higher incidence of hepatic adverse events reported with nevirapine. 21,30,31 The majority of hepatic effects observed with nevirapine are mild in severity, although serious and sometimes fatal cases of hepatotoxicity have been reported. 21,24,32
In Phase IIb and III trials, no clinically relevant significant hepatotoxicity has been observed in patients treated with ETR-based regimens when compared with patients receiving placebo/control. 7 –11,13,14 The incidence of hepatobiliary events has been shown to be low. Reported hepatic events have been mild to moderate in severity.
No consistent or frequent hepatic adverse events were observed in the TMC125-C203 study and no clinically relevant difference was noted versus placebo in the incidence of hepatobiliary disorders (3.4% with ETR vs. 6.1% with placebo) or related laboratory abnormalities. 9 A low number of grade 3 or 4 treatment-emergent changes in hepatic parameters was observed (<2%). No patients discontinued treatment as a result of hepatic events.
Likewise, pooled 48-week DUET data revealed a similar incidence of hepatic adverse events (any severity) in patients receiving ETR relative to placebo (6.5% vs. 6.1% for ETR and placebo, respectively; Table 2). 11 Grade 3 or 4 hepatic events were infrequent and showed no difference in incidence across treatment groups (3.0% in both the ETR and placebo groups). The incidence of hepatic transaminase abnormalities [alanine aminotransferase (ALT) or aspartate aminotransferase (AST) elevation] did not differ across groups and there was no clinically relevant mean change from baseline for ALT and AST in either treatment group. Discontinuation due to hepatic abnormalities was low in both treatment groups (1.0% with ETR and 0.7% with placebo).
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BR, background regimen of darunavir/ritonavir, investigator-selected NRTIs. Enfuvirtide use was optional; SAE, serious adverse event. N = number of patients, n = number of patients with observations.
DUET subgroup analyses in hepatitis B and/or C coinfected patients have shown a similar incidence of hepatic adverse events when compared to placebo, irrespective of hepatitis B and/or C coinfection status, 33 suggesting that ETR-treated patients with hepatitis coinfection are not at increased risk of hepatic adverse events related to ETR. Population pharmacokinetic analysis of the DUET trials showed reduced clearance for ETR in HIV-1-infected patients with hepatitis C coinfection, and hepatitis B and/or C coinfection was associated with a 1.35-fold increase in ETR exposure (p = 0.0028). 34 However, the incidence and severity of adverse events were similar in patients with and without hepatitis B and/or C coinfection. No dose adjustment is necessary in patients coinfected with hepatitis B and/or C and standard clinical monitoring of these patients is considered adequate. Overall, ETR does not appear to be associated with liver toxicity and may therefore be considered for patients at risk of hepatic adverse events. The pharmacokinetics of ETR have not yet been evaluated in patients with severe hepatic impairment (Child–Pugh C); therefore its use cannot be recommended in this patient subpopulation. 35,36
Dyslipidemia
Dyslipidemia is an important clinically relevant adverse event of combination HIV therapy. 37 –40 The clinical implications of increased serum concentrations of lipids and lipoproteins have been highlighted in the literature. 39,41 In approximately 70% of cases, antiretroviral-induced dyslipidemia [particularly increases in low-density lipoprotein (LDL) cholesterol and triglycerides] may result in an increased risk of coronary heart disease. 40
Dyslipidemia may be manifested by elevation of total cholesterol, LDL cholesterol, and triglyceride concentrations, or by a decrease in the cardioprotective high-density lipoprotein (HDL) cholesterol concentration in serum. The nature and extent of the antiretroviral-induced changes in the concentrations of lipids or lipoproteins in the blood vary, but NNRTI-based regimens generally produce fewer changes than protease inhibitors (PIs). 37,42 –45 The role of NNRTIs in the development of metabolic complications leading to conditions such as dyslipidemia and hypertriglyceridemia varies with the specific NNRTI chosen, and is controversial. 21,46 –51
While efavirenz has been shown to have the potential to induce hyperlipidemia, nevirapine has conversely been reported to lower serum lipid concentrations. 20,32,37,50,51 Data from the 2NN lipid substudy showed a greater increase in levels of the cardioprotective HDL cholesterol and a lower increase in triglycerides and LDL cholesterol in patients receiving nevirapine compared with those receiving efavirenz. 51
Although the effect of ETR on lipid levels is not known for treatment-naive patients, there is substantial evidence from clinical trials that demonstrates that the addition of ETR does not increase dyslipidemia in treatment-experienced patients. Data from Phase IIb and III clinical trials provide no evidence for a clinically relevant effect of ETR on plasma lipid concentrations. 7 –11 In the TMC125-C203 study, no important treatment differences compared with placebo were observed in the incidences of individual laboratory abnormalities, including concentrations of triglycerides and cholesterol. 9 While grade 3 or 4 adverse events included a low incidence of hypertriglyceridemia, the frequency did not differ across treatment groups (3.4% for ETR groups vs. 3.0% in placebo plus background regimen groups). In the Phase IIb TMC125-C227 study, grade 3 or 4 lipid abnormalities (any cause) were found to be less frequent with ETR than with control PI therapy. 52
Pooled 48-week DUET data confirm these findings and demonstrate that lipid levels in ETR-treated patients are generally comparable to placebo (Fig. 3). 11 A small number of patients randomized to ETR reported grade 3 or 4 changes in total cholesterol (8.1% for grade 3 vs. 5.3% with placebo; no grade 4), triglycerides (5.7% vs. 4.0% and 3.5% vs. 1.8% for grade 3 and grade 4, respectively), or LDL cholesterol (7.2% vs. 6.6% with placebo; no grade 4) concentrations, but no significant differences were observed when compared with placebo groups. Any small changes observed were not considered to be clinically relevant. It is worth noting that most of the lipid data for ETR were generated when the drug was combined with a background regimen containing a ritonavir-boosted PI, which may affect plasma lipid levels to varying degrees.

Mean change from baseline in (
While these findings indicate that ETR does not adversely affect plasma lipid concentrations when included in antiretroviral regimens, further studies are required to confirm the effects of the drug on lipid levels, particularly in patients with preexisting dyslipidemia and other cardiovascular risk factors.
Use in pregnancy
The teratogenic potential of the first generation NNRTIs varies with the individual drugs. Efavirenz use has been limited in women of a child-bearing age due to retrospective reports of neural tube defects (including meningomyelocele) in babies whose mothers were exposed to efavirenz-containing regimens during the first trimester of pregnancy. 19,53 Although a causal relationship of these events to efavirenz treatment has not been established, these data have resulted in the recommendation that an alternative agent be used for women in the first trimester of pregnancy. 16,17,54 On the other hand, data from the Antiretroviral Pregnancy Registry do not suggest any increase in birth defects following nevirapine use. 55 Consequently, nevirapine is widely used in pregnancy as a component of highly active antiretroviral therapy and is recommended for use in the prevention of mother-to-child HIV transmission. 55,56
No recommendations can currently be made for the use of ETR in pregnant women due to insufficient clinical data. 35 However, data from reproduction studies undertaken in rats and rabbits, at systemic exposures equivalent to those observed in humans at the recommended clinical dose, suggest that ETR has no direct or indirect teratogenic effects with regard to pregnancy, embryonal/fetal development, parturition, or postnatal development. 57 Additionally, an assessment of pharmacokinetics and safety of ETR in five pregnant HIV-infected women showed comparable ETR pharmacokinetics to nonpregnant adults and no effect of ETR on fetal or neonatal toxicity. 58 To date, there has been only one reported case of a pregnant woman with multiclass HIV resistance who was successfully treated with a regimen composed of ETR, darunavir, enfuvirtide, and tenofovir/emtricitabine. 59
Combining ETR with other antiretroviral classes
The safety and tolerability profile of ETR differs from that of most PIs, with no apparent association with gastrointestinal events, 54 hyperbilirubinemia, hyperlipidemia, and hyperglycemia. Because of this nonoverlapping safety profile, ETR would appear to be a good candidate for combined treatment with PIs. Indeed, Phase IIb and Phase III studies have combined ETR with ritonavir-boosted darunavir 7,8,15 or boosted lopinavir 10 and investigator-selected nucleoside reverse transcriptase inhibitors, and showed that the addition of ETR to boosted PI-based regimens does not lead to further reported toxicity, except for rash (as discussed previously).
Similarly, there is no substantial overlap in the safety and tolerability profile of ETR and other antiretroviral agents such as the integrase inhibitor raltegravir, 15,60 the CCR5 antagonist maraviroc, 61 and the fusion inhibitor enfuvirtide, 62 suggesting that ETR would be well tolerated in combination with these therapies. Pharmacokinetic data indicate that ETR can be combined with enfuvirtide and raltegravir without dose adjustment. 63 Pharmacokinetic data also show that ETR can be coadministered with maraviroc, but a dose adjustment of maraviroc is required. 64
Discussion
Based on the data from more than 1000 HIV-infected, treatment-experienced patients who received ETR in Phase IIb and III trials, ETR has been shown to be well tolerated and not associated with any particular signature safety issues, other than rash. Notably, the safety and tolerability of ETR in the Phase III DUET trials were generally similar to placebo. The only adverse event that was more common with ETR was rash. When rash does occur with ETR, it is generally mild to moderate, manageable, resolves with continued treatment, and infrequently results in interruption or discontinuation of therapy. Moreover, studies have shown that patients with previous NNRTI-related rash were not predisposed to develop rash on initiation of ETR in their regimen. The tolerability profile of ETR appears to represent an improvement relative to earlier NNRTIs, as it is not associated with increased neuropsychiatric, lipid, or hepatic adverse events.
In clinical development, ETR has shown durable efficacy and, with the exception of rash, a safety profile similar to placebo. Furthermore, the nature and magnitude of adverse events observed with ETR suggest that the drug may offer improved tolerability over first-generation NNRTIs. In addition ETR could offer safety benefits over first-generation NNRTIs in early experienced patients. ETR can be used in combination with other antiretrovirals such as boosted PIs and raltegravir with little potential for additional toxicity. Therefore, given this favorable safety profile along with its enhanced barrier to the development of resistance and demonstrated efficacy, ETR provides an important treatment option for HIV-infected, treatment-experienced patients. The safety of ETR in treatment-naive patients is being explored in ongoing clinical trials. New trials are also investigating the safety and tolerability of ETR in treatment-experienced children and adolescents.
Footnotes
Acknowledgments
We are grateful to the patients, investigators, study center staff, Data and Safety Monitoring Boards, and Tibotec study personnel involved in the etravirine clinical development program. We thank Benny Baeten, Andrew Clark, and James Witek from Tibotec for their important contributions to the manuscript. We also thank Louise Marks (Gardiner-Caldwell Communications, Macclesfield, UK) for her assistance in drafting the manuscript and collating author contributions. This service was funded by Tibotec BVBA, Mechelen, Belgium.
Author Disclosure Statement
Giovanni Di Perri has received financial support from BMS, GSK, Abbott, Roche, Tibotec, Pfizer, Gilead, Astra Zeneca, and Merck Sharp & Dohme. William Towner has received research grant support from Tibotec, Merck, Pfizer, BMS, Gilead, and Schering-Plough. Brian Woodfall, Goedele De Smedt, and Monika Peeters are full-time employees of Tibotec, the developer of etravirine.