
Abstract
Sustained viral suppression with antiretroviral therapy improves clinical outcomes for HIV-infected individuals. Study M05-730 evaluated the long-term antiviral activity, safety, tolerability, emergence of resistance, and compliance with once-daily (QD) versus twice-daily (BID) lopinavir/ritonavir (LPV/r) combination therapy in treatment-naïve, HIV-1-infected subjects through 96 weeks. Antiretroviral-naïve subjects with HIV-1 RNA levels >1000 copies/ml were randomized to LPV/r QD (N = 333) or BID (N = 331) with tenofovir DF and emtricitabine. Through 96 weeks, 77 subjects from each group discontinued prematurely; adverse or HIV-related events contributed to discontinuation of 36 subjects overall, with no significant differences between treatment groups. At 96 weeks, 216 QD subjects (64.9%) and 229 BID subjects (69.2%) had HIV-1 RNA <50 copies/ml (p = 0.249) by intent-to-treat analysis. Evaluation of the time to virologic failure indicated that 85.0% and 80.7% of QD and BID subjects, respectively, maintained virologic suppression through 96 weeks (p = 0.638). QD subjects demonstrated greater adherence levels. There were no significant differences in virologic response when subjects were analyzed according to baseline disease state. Emergence of postbaseline resistance mutations occurred at similar low rates in each dosing group. Diarrhea was the most common moderate-to-severe drug-related adverse event reported; the most common Grade 3+ laboratory abnormalities were elevations of total cholesterol and triglycerides, occurring with similar incidence regardless of LPV/r dosing frequency. QD dosing of LPV/r was associated with similar durability of viral suppression and low rates of genotypic resistance and treatment-limiting adverse events as compared with BID dosing in treatment-naïve subjects through 96 weeks of treatment.
L
Study M05-730 was an open-label, randomized, multicountry Phase 3 trial designed to compare the safety, tolerability, and antiviral efficacy of LPV/r administered as the tablet formulation dosed once versus twice daily in combination antiretroviral therapy in treatment-naïve, HIV-1-infected subjects. The primary intent-to-treat, noncompleter =failure (ITT, NC = F) efficacy results at 48 weeks have previously been described and demonstrated noninferior antiviral activity of LPV/r dosed QD compared with BID, with 77% of QD-treated and 76% of BID-treated subjects achieving HIV-1 RNA <50 copies/ml at this time point. 7 The objectives of this final analysis include a comparison of the maintenance of antiviral activity for QD and BID LPV/r using intent-to-treat and time-to-loss of virologic response analyses, and an evaluation of the long-term treatment-emergent adverse events and resistance. This report describes the efficacy, safety, and antiretroviral resistance associated with QD or BID LPV/r dosing through 96 weeks of treatment, as well as treatment compliance through the first 12 weeks of treatment.
The design of Study M05-730 has been previously described in detail. 7 Antiretroviral-naïve, HIV-1-infected adult subjects with HIV-1 plasma RNA levels ≥1000 copies/ml and any CD4+ T-cell count were eligible to participate. All subjects receiving at least one dose of study medication were included in the analysis. HIV-1 plasma RNA, CD4+ T-cell counts, adverse events, and laboratory values were measured as previously described. 7
The primary 96-week efficacy endpoint was the proportion of subjects with plasma HIV-1 RNA <50 copies/ml according to an intent-to-treat analysis with noncompleters considered failures (ITT, NC = F). The proportion of subjects with HIV-1 RNA <50 copies/ml was also computed using an on-treatment analysis, where subjects with missing data were excluded from analysis. Differences between treatment groups were assessed using Fisher's exact test, and 95% confidence intervals for the differences in response rates were calculated. The time to loss of virologic response was summarized using Kaplan–Meier methodology; the Cox proportional hazards model was used to assess differences between dosing groups. The time to loss of virologic response was determined as the first of the following: day 1, if two consecutive plasma HIV-1 RNA levels <50 copies/ml were never observed; the first of two consecutive visits with plasma HIV-1 RNA levels ≥50 copies/ml for subjects with confirmed HIV-1 suppression; and the day of the final plasma HIV-1 RNA measurement, if it was ≥50 copies/ml.
Changes from baseline in CD4+ T-cell counts and lipid parameters were analyzed using one-way ANOVA. Fisher's exact test was used to compare the treatment groups with respect to incidence of adverse events and Grade 3 or greater laboratory abnormalities.
Adherence to LPV/r was assessed through the first 12 weeks of treatment using Medication Event Monitoring System (MEMS) pill bottle caps (Advanced Analytical Research on Drug Exposure, Ltd., Zurich, Switzerland). Three measures of adherence to LPV/r were calculated as described in earlier studies 5,6 : “Taking Compliance” is the percentage of LPV/r doses taken relative to those prescribed, “Correct Dosing Compliance” is the percentage of days on which the correct number of LPV/r doses was taken, and “Timing Compliance” is the percentage of LPV/r doses taken within ±3 h of the prescribed time interval. Differences in the distribution of subjects' adherence were compared between groups using the Wilcoxon rank sum test.
Genotypic resistance testing was performed for subjects with a confirmed HIV-1 RNA rebound ≥50 copies/ml where the confirmation sample was >400 copies/ml. Additional genotypic resistance testing was performed for subjects who had at least one HIV-1 RNA level >400 copies/ml after week 24 and a final value ≥50 copies/ml; the sample corresponding to the date of the last HIV-1 RNA value >400 copies/ml was used for resistance testing.
The study was approved by local institutional ethics review panels and subjects provided signed informed consent to participate. The trial was registered with
Six hundred and sixty-four subjects were randomized and treated: 333 in the LPV/r QD treatment group and 331 in the LPV/r BID treatment group. The study arms had similar baseline demographics; however, subjects in the BID group had a higher mean HIV-1 RNA level at baseline (5.05 log10 copies/ml) compared to subjects in the QD group (4.93 log10 copies/ml, p = 0.020), and a greater proportion of BID subjects had baseline HIV-1 ≥5 log10 copies/ml (58.3%) relative to QD subjects (48.0%, p = 0.008).
There were 77 discontinuations through 96 weeks from each treatment group, and no statistically significant differences between regimens in the overall proportion of subjects discontinuing prematurely or for any indicated reason. The majority of premature treatment discontinuations (104/154, 67.5%) occurred during the first 48 weeks of treatment. The most common reason for treatment discontinuation after week 48 was loss to follow-up (15/50, 30.0%). Treatment-emergent adverse events contributed to discontinuation of 20 QD subjects (6.0%) and 14 BID subjects (4.2%); the types of adverse events contributing to discontinuation were similar between groups. Ten discontinuations (four QD subjects and six BID subjects) occurring after week 48 were due to adverse events; thus the majority of discontinuations related to adverse events occurred before week 48.
At week 96, there were no significant differences between the proportions of QD- and BID-treated subjects with HIV-1 RNA <50 copies/ml (or <400 copies/ml) by ITT, NC = F analysis or on-treatment analysis (Fig. 1A–C). The difference between the QD and BID mean ITT response rates was −4.3% [95% confidence interval (CI), −11.5% to 2.8%]. Kaplan–Meier analysis of the time-to-loss of virologic response revealed that similar proportions of QD subjects and BID subjects maintained virologic suppression at a threshold of 50 copies/ml at 96 weeks (Fig. 1D).
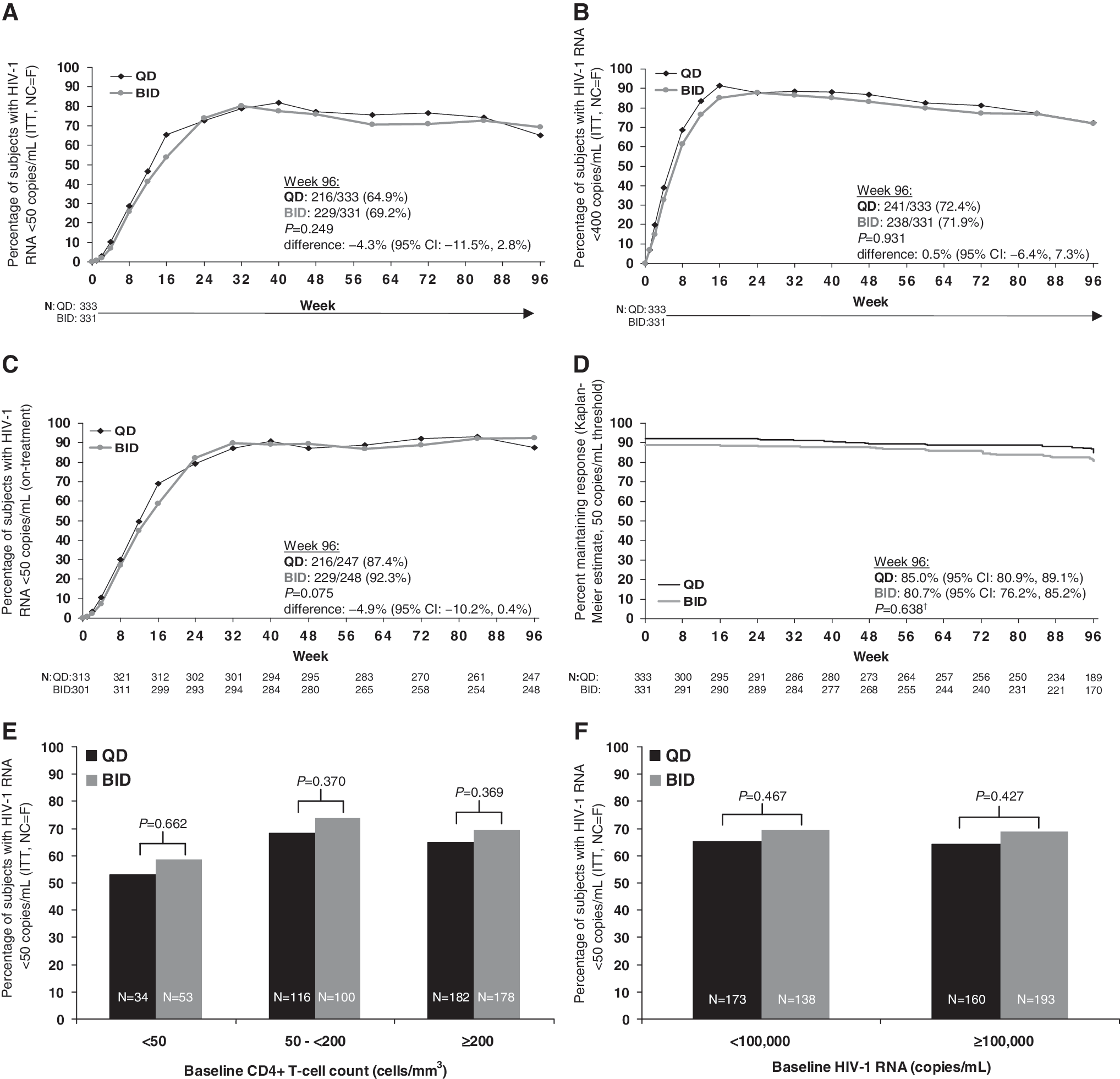
Antiviral efficacy. Insets (
Instances of intermittent viremia, defined as an HIV-1 RNA level ≥50 copies/ml after week 24 followed by an HIV-1 RNA measurement <50 copies/ml at the subsequent visit, occurred with similar frequency in both treatment groups. There were 91 such instances in 76 QD-treated subjects and 97 instances in 83 BID-treated subjects. Of the individuals who exhibited their first rebound of HIV-1 RNA ≥50 copies/ml at week 96, 16 of 22 QD subjects (72.7%) and 5 of 8 BID subjects (62.5%) had HIV-1 RNA <200 copies/ml.
When subjects were analyzed according to baseline HIV-1 RNA or baseline CD4+ T-cell count, ITT response rates at week 96 were similar between treatment groups within each of the subgroups examined (Fig. 1E–F).
Mean increases in CD4+ T-cell counts from baseline through 96 weeks were similar between groups (+238.4 cells/mm3 for QD and +254.0 cells/mm3 for BID, p = 0.269).
Subject adherence was significantly better in the LPV/r QD-dosed group by all measures of adherence through week 12, the period in which MEMS cap monitoring was employed. Mean Taking Compliance was 99% for QD-treated subjects and 93% for BID-treated subjects (p < 0.001). The mean Correct Dosing Compliance was 93% and 81% for LPV/r QD and BID subjects, respectively (p < 0.001); lastly, the mean Timing Compliance was 80% in the QD-dosed group and 67% for the BID-dosed group (p < 0.001).
The frequencies of specific treatment-emergent, study drug-related adverse events of at least moderate severity were similar between treatment arms (Table 1). Gastrointestinal adverse events were most commonly reported, including diarrhea, nausea, and vomiting. Adverse events of diarrhea contributed to premature study drug discontinuation for 10 (3.0%) subjects taking LPV/r QD and 6 (1.8%) BID-dosed subjects (p > 0.100).
QD N = 330, BID N = 327.
QD N = 330, BID N = 326.
BID N = 327.
BID N = 328.
The incidence of laboratory abnormalities of Grade 3 or greater was not significantly different between subjects in the QD and BID treatment groups (Table 1). Elevations of total cholesterol, triglycerides, and lipase were the only Grade 3+ abnormalities observed in ≥5.0% of subjects in either treatment group. There were statistically significant differences between QD and BID groups for the following lipid parameters; otherwise, no differences for lipid parameters were noted: mean increases in triglyceride levels [QD: +0.448 mmol/liter (+39.7 mg/dl), N = 257; BID: +0.719 mmol/liter (+63.7 mg/dl), N = 253; p = 0.048]; mean increases in non-HDL cholesterol levels [QD: +0.669 mmol/liter (+25.7 mg/dl), N = 256; BID: +0.824 mmol/liter (+31.7 mg/dl), N = 253; p = 0.050]; and mean decreases in LDL:HDL ratio (QD: −0.377, N = 252; BID: −0.082, N = 251; p = 0.029).
Postbaseline genotypic resistance data were available for 25 QD-treated subjects and 26 BID-treated subjects. The M184V mutation, conferring resistance to emtricitabine and lamivudine, was observed in 4 of 25 QD subjects (16.0%) and 6 of 26 BID subjects (23.1%). There was no evidence of development of resistance to TDF. One BID-treated subject who had extensive protease mutations at baseline developed additional primary lopinavir-associated resistance mutations (I47V and I50V). The incidence of new resistance mutations was not significantly different between the QD and BID treatment groups for any study drug.
The findings from this week 96 analysis support and extend the previously published 48-week results from this study, 7 confirming that LPV/r dosed QD has similar antiviral efficacy, tolerability, and selection of antiviral resistance compared to BID LPV/r in treatment-naïve subjects.
At 96 weeks, comparable rates of QD (65%) and BID (69%) subjects had HIV-1 RNA levels <50 copies/ml by intent-to-treat analysis. On-treatment response rates at week 96 were also similar between the LPV/r QD and BID dosing groups (87% and 92%, respectively). The durability of QD and BID LPV/r dosing in this population was evaluated by time to loss of virologic response, which revealed that a similar proportion of subjects in each group maintained virologic suppression through 96 weeks. Furthermore, virologic responses through 96 weeks in subgroups defined by baseline HIV-1 RNA and CD4+ T-cell count were similar between the two dosing groups. These findings suggest that the durability of antiviral activity with QD LPV/r dosing is analogous to that seen with BID dosing. This conclusion is further supported by the similar frequency of intermittent viremia and infrequent emergence of resistance mutations in both treatment groups.
As reported previously, QD dosing of LPV/r was associated with better treatment compliance than BID. 6,8,9 However, overall adherence was high in both groups, possibly explaining the absence of a difference in virologic response rates. It has been estimated that a Taking Compliance rate of 85% is necessary to achieve suppression of HIV-1 RNA levels to <50 copies/ml, 10 a threshold that was exceeded in both treatment groups in this study.
Through 96 weeks, once-daily dosing of LPV/r continued to show tolerability comparable to that of twice-daily dosing, as evidenced by similar low rates of study drug discontinuation due to adverse events and of premature study discontinuation in general. Overall safety was also similar between the two dose groups through 96 weeks, as measured by the frequency of adverse events and laboratory abnormalities. Consistent with the known safety profile of LPV/r, diarrhea was the most commonly reported moderate-to-severe study drug-related adverse event, occurring at a similar rate in both dosing groups. The similar incidence of diarrhea leading to drug discontinuation suggests that there was no difference in the frequency or severity of diarrhea between treatment groups in this study. This finding contrasts with the results of a previous smaller trial of LPV/r, M02-418, in which LPV/r was administered as a soft gel capsule dosed once vs. twice daily. In that study a significant difference in rates of diarrhea was observed between subjects dosed once daily and those dosed twice daily. 6
Consistent with previous studies of QD and BID LPV/r in antiretroviral-naïve subjects, 5,6 statistically significant changes in laboratory values were deemed to be of limited clinical significance, as lipid elevations resulted in study discontinuation for only one subject from each treatment group and increased lipase levels did not lead to adverse events of pancreatitis for any subject.
Limitations of this study include the open-label study design, which might have led to bias in the reporting or management of adverse events. However, blinding to treatment allocation was not practical and would have obviated the ability to assess adherence as a function of dosing frequency. Another limitation is that treatment compliance was monitored only through the first 12 weeks of dosing; long-term adherence has previously been compared for QD and BID dosing of LPV/r through 96 weeks and showed similar results in terms of rate and pattern of adherence. 6 Lastly, these results cannot be generalized to patients who might have altered LPV pharmacokinetic profiles, such as pregnant women, pediatric populations, or patients taking drugs that alter LPV/r metabolism.
In conclusion, these findings confirm and extend the 48-week results from this study demonstrating similar efficacy, safety, and low rate of genotypic resistance for LPV/r dosed once daily or twice daily in treatment-naïve, HIV-1-infected individuals. QD LPV/r administration is therapeutically equivalent to BID dosing and may represent an important therapeutic option for patients who would benefit from a simplified treatment regimen.
Footnotes
Acknowledgments
The authors are grateful to all trial subjects and researchers who contributed to this study; a list of participating investigators is provided in Gathe et al. 7 Abbott provided design and financial support of Study M05-730. The authors acknowledge Elaine M. Smith, Ph.D. (senior medical writer, Abbott) for assistance in writing and editing the manuscript.
Author Disclosure Statement
D. Cohen, L. Fredrick, C. Naylor, B. da Silva, and B. Bernstein are employees of Abbott and may hold Abbott stock or options. J. Gonzalez-Garcia has received advisory fees from Gilead Sciences and Abbott; M. Johnson has received speaker, advisory, and/or investigator fees from Abbott, BMS, MSD, GSK, and Pfizer.