
Abstract
Mutations in the env gene of HIV-1 have been the primary focus in most epidemiologically related cohort studies of virus evolution and very limited studies have focused on the reverse transcriptase (RT) region, the primary target of antiretroviral therapy (ART). Hence, we measured the selection pressure and searched for the positively selected sites in the RT sequences amplified from HIV-1-infected heterosexual transmission pairs. Married couples (n = 10) who were ART naive were included in this study. Phylogenetic analysis, the measurement of synonymous and nonsynonymous ratio (dN/dS) and the interpatient nucleotide variation, was done. Phylogenetic analysis demonstrated distinct subclusters of the RT sequences from heterosexual transmission pairs and the median (IQR) nucleotide variation between the epidemiologically related transmission pairs was significantly (p < 0.001) lower [0.01% (0.01–0.02%)] compared to the epidemiologically unrelated transmission pairs [0.04% (0.03–0.04%)]. The ratio of dN/dS was <1 and codons 135, 162, 166, 207, and 211 were positively selected in >50% of the donor and recipient RT sequences. Purifying selection pressure and low nucleotide variation in the RT sequences between epidemiologically related transmission pairs highlight its essential role in HIV-1 replication. The effect of the RT positively selected mutations that persist over time following transmission between individuals needs to be studied to determine the fitness cost of the mutations in vivo, which may possibly represent good targets for inclusion in HIV-1 vaccines.
O
Retrospective HIV-seropositive samples that were collected from ART-naive transmission pairs (married couples, n = 10), during their visit to YRG CARE Medical Centre, were obtained to diagnose HIV. Follow-up samples were also collected after a period of 10–12 months from 12 subjects. Women (recipient) included in the study self-reported to have acquired HIV-1 infection through their husbands (donor). The mean (standard deviation) age of males and females was 34 ± 8.8 and 29 ± 8.6 years, respectively, and their median (interquartile range, IQR) CD4+ T cell count was 386 (295–595) and 437 (366–751) cells/μl. The study protocol was approved by YRG CARE's Institutional Review Board and written informed consent was obtained from all the participants included in the study.
HIV-1 RNA was isolated using the QIAamp viral RNA kit (QIAGEN, Inc., USA). The RT region (20–240) was amplified from cDNA using nested PCR as described earlier 19 with appropriate controls. Bidirectional population sequencing of purified products was done using ABI 3100-Avant genetic analyzer (Applied Biosystems, USA). Sequences were edited using Seqscape (Applied Biosystems, USA, v. 2.5) software. Sequence alignment was carried out on the translated amino acid sequence in Clustal W, 20 as implemented in MEGA version 3.1. 21 Maximum likelihood, minimum evolution, neighbor-joining phylogenetic analyses were used to explore the heterosexual relationships among the donor and recipient RT sequences. The robustness of each tree was evaluated by bootstrap analysis of 1000 replicas.
Interpatient nucleotide distance was measured by comparing the donor and recipient RT sequences using the Kimura two-parameter nucleotide distance method as implemented in MEGA version 3.1.
21
The RT sequences amplified from primary and follow-up samples were classified into distinct groups as donor and the recipient and compared with the consensus C reference sequence to calculate the ratio of nonsynonymous and synonymous substitutions (dN/dS) and the analysis was done using Syn-SCAN.
22
Drug resistance mutations were identified using the Stanford HIV-1 Drug Resistance Database (
The phylogenetic analysis demonstrated distinct subclusters of the RT sequences from heterosexual transmission pairs (Fig. 1). The median (IQR) nucleotide distance between the epidemiologically related transmission pairs was significantly (p < 0.001) lower [0.01% (0.01–0.02)] than the epidemiologically unrelated transmission pairs [0.04% (0.03–0.04)]. A previous study related the level of nucleotide variation among epidemiologically linked mother and infant transmission pairs with viral pathogenesis. 18 Two transmission pairs (Pair 2 and 8) showed no sequence variation (0.00%), however, clustered separately, confirming that they belong to the transmission chain. The remaining transmission pairs exhibited nucleotide distances ranging from 0.01% to 0.02%. The extent of HIV-1 nucleotide variation observed between the transmission pairs could be associated with duration of infection, selective pressure imposed by the host immune system, and the rate of disease progression. 24 –28 Owing to the cross-sectional analysis and lack of information on the duration of HIV-1 infection, these aspects could not be delineated from the level of nucleotide variation.
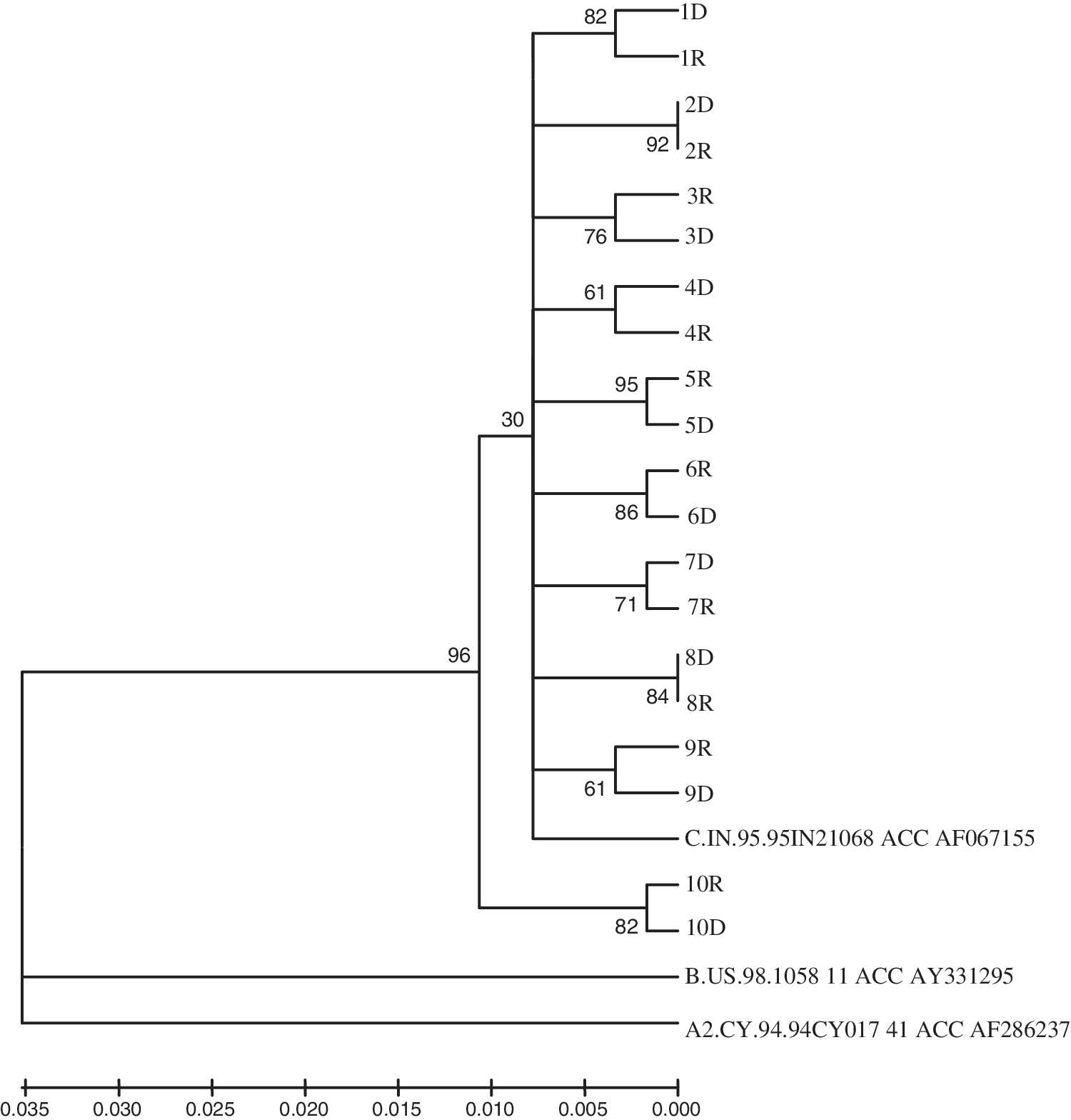
Phylogenetic analysis of HIV-1 RT sequences from heterosexual transmission pairs (D, donor; R, recipient) along with the consensus reference sequence (HIV-1 subtypes A, B, and C) downloaded from the Los Alamos National Laboratory HIV Molecular Immunology Database. The neighbor joining tree is based on the distance calculated between the nucleotide sequences from the transmission pairs. The numbers on the branch points indicate the percent occurrence of branches over 1000 bootstrap resamplings of the data set.
The ratio of dN/dS in the donor [0.2 (0.1–0.2)] and recipient sequences [0.2 (0.12–0.23)] was <1, which indicates the purifying selection pressure 29 over the RT region in parallel with other studies. 30 –32 . Selective transmission of a few viral variants has been proposed in horizontal as well as vertical transmission due to the detection of highly homogeneous env sequences in recently infected individuals and due to the detection of greater env gene diversity in chronically infected individuals. 33 –38 The higher dN/dS estimates have been linked to selection for changes in the virus by the host adaptive immune response, which includes neutralizing antibodies, cytotoxic T cells, and/or T helper cell responses. 39,40 These changes will enable the virus to escape from the immune response and are most likely to be associated with longer survival times. 27,41 However, synonymous nucleotide changes were usually observed to predominate among the surviving variant sequences and dN/dS tends to be <1 when there is a requirement to maintain protein function. 30 –32
No drug resistance mutations were observed in the RT sequences amplified from the transmission pairs. Positive selection of codons 135, 162, 166, 207, and 211 occurred in >50% of the donor and the recipient RT sequences (Fig. 2). These codons were positively selected in the 12 sequences collected during follow-up, of which eight were from the transmission pairs (n = 4). Codons positively selected in the donor sequences were also observed to be positively selected in the recipient sequences. Repeated positive selection of the same amino acid mutations that are located in the regions of the cytotoxic T lymphocyte epitope (aa 128–135, 158–166, and 202–212) is likely to be a characteristic of the epitope subjected to the strongest host immune selective pressure. 9,42 –47 Baseline polymorphisms in treatment-naive subjects within RT have been linked to resistance to RT inhibitors. 30,48 –50 It appears that CD8+ T cell escape variants could become fixed and propagated through populations 47 in the same way viruses resistant to ART are transmitted. 51
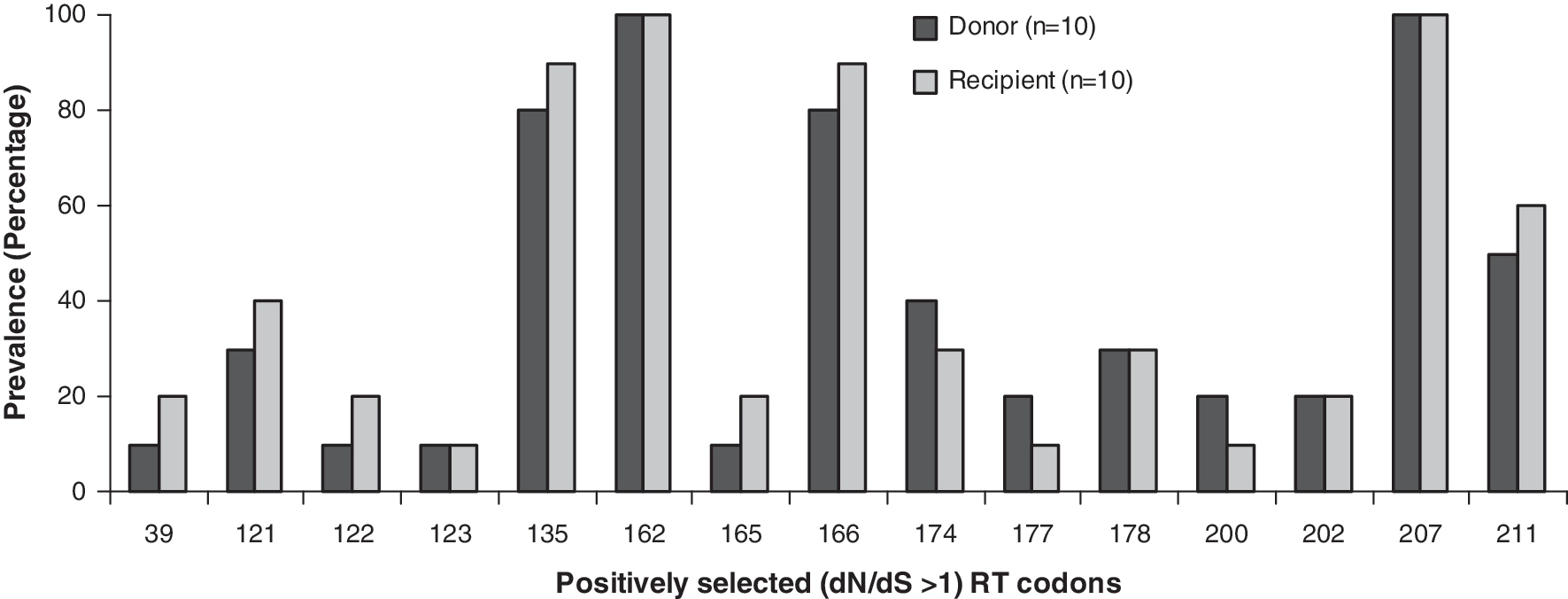
Occurrence of positively selected sites in HIV-1 RT among the transmission pairs. The dark and light colored bars represent the prevalence of positively selected codons in the RT sequences amplified from the donor and the recipient, respectively.
An earlier study 52 proposed two rationales for the lack of reversion of CD8+ T cell escape variants: the absence of a significant influence on viral fitness and the existence of CD8+ T cell-mediated selective pressure restricted by a human leukocyte antigen (HLA) allele. Since no evidence was reported to support the above affirmed rationale, future studies are required to identify associations between an HLA allele and specific amino acid polymorphism and also to evaluate dominant immune responses to the antigenic peptides of subtype C pol, which will be needed to gain insight into the viral pathogenic mechanisms as well as the design of a vaccine.
In conclusion, the observed purifying selection pressure and low RT nucleotide variation between epidemiologically related transmission pairs highlight its essential role in HIV-1 replication. The effect of RT positively selected mutations that persist over time following transmission between individuals needs to be studied to determine the fitness cost of the mutations in vivo, which may possibly represent good targets for inclusion in HIV-1 vaccines.
Nucleotide Sequence Accession Numbers
The GenBank accession numbers of the HIV-1 RT described here are EU429988 through EU430023 and EU597182 through EU597197.
Footnotes
Author Disclosure Statement
No competing financial interests exist.