
Abstract

E
More precisely, we assessed changes in innate cell numbers and function during TIs in fresh blood samples from an observational cohort of 14 chronically HIV-1-infected patients (age ≥18 years) on suppressive ART (plasma HIV-1 RNA <50 copies/ml). ART initiation for all patients ranged from 1986 to 2000. All patients underwent two consecutive TIs of 6 and 18 weeks, with intervening ART resumption and resuppression to <50 copies HIV mRNA/ml for 4 weeks. All parameters were assessed at the start of each TI as well as at weeks 4 and 6 of the first TI, and weeks 4, 6 and 18 of the second TI. Informed consent was obtained according to the Human Experimentation Guidelines of the U.S. Department of Health and Human Services and of the authors' Institutions. The study protocol was approved by the Institutional Review Boards of the Wistar Institute and Philadelphia FIGHT.
Immunophenotyping characterization of NK, DC [plasmacytoid (PDC) and myeloid (MDC)], and T cell subsets was performed by same day whole blood four-color staining as previously described 7 by using the following combinations of directly fluorochrome-conjugated antihuman cell surface monoclonal antibodies from Becton Dickinson (BD) Biosciences (San Diego, CA): (1) CD56-fluorescein isothiocyanate (FITC)/CD16-phycoerythrin (PE)/CD3-PerCP/CD161-allophycocyanin (APC) (NK cells), (2) CD56-FITC/CD161-PE/CD3-PerCP/HLADR-APC (activated NK cells), (3) Lin-1 (CD3/CD14/CD16/CD19/CD20/CD56)-FITC/CD123-PE/HLA-DR-PerCP/HLA-ABC-APC (PDC), (4) Lin-1-FITC/CD11c-PE/HLA-DR-PerCP/HLA-ABC-APC (MDC), and (5) CD28-FITC/CD4-PE/CD3-PerCP/CD38-APC (T cells). Definitions of total NK cells included all CD3−/CD161+/–/CD56+/–/CD16+/– NK subsets (with the exception of CD3−/CD161−/CD56−/CD16+/– NK subsets, which were not included) whereas definitions of total HLA-DR+ NK cells included all CD3−/HLA-DR+/CD56+/–/CD161+/–subsets (with the exception of CD3−/HLA-DR+/CD56−/CD161−, which were not included)]. PDC and MDC were defined as Lin-1−/HLA-DR+/CD123+ and Lin-1−/HLA-DR+/CD11c+, respectively, whereas CD8 cells were defined by gating on CD3+/CD4− events. Results were expressed as mean fluorescent intensity (MFI), percent positive (%), and cells/mm3. In the text cell numbers represent cells/mm3.
PDC function within fresh peripheral blood mononuclear cell (PBMC) preparations was assessed by measurement of spontaneous (no stimulation), CpG oligodeoxynucleotide class A (CpG-2216, 10 μg/ml, Integrated DNA Technologies, Coralville, IA), or heat-inactivated PR8 influenza virus (10 HA units/ml)-induced IFN-α levels following 18 h of culture. Cell-free supernatants were harvested and tested in duplicate for IFN-α using commercial ELISA plates (PBL Biomedical Laboratories, Piscataway, NJ). Sensitivity of the assay was approximately 9.7 pg/ml.
NK function was assessed measuring spontaneous (no stimulation) and CpG-2216 (10 μg/ml)-induced NK cell-mediated cytotoxicity by standard 51Cr release assay, as previously described, using fresh PBMC preparations as effectors cells against the tumor derived erythroblastoid cell line K562. 8 Results were expressed as area under the curve (AUC) for effector:target ratios of 50:1, 25:1, 12:1, and 6:1 for both spontaneous and induced NK function.
All statistical tests were performed using GraphPad Prism4 software. Briefly, a one-way ANOVA (Kruskal–Wallis test) was performed followed by Dunn's multiple comparison test. Differences were considered significant if p < 0.05. Short-term viremia following TIs did not significantly (p > 0.05) affect the number of CD4+ T cells, although as expected clear changes in the activation state of both CD4+ and CD8+ T cells were noted as indicated by cell surface CD38 expression (Fig. 1A) and numbers of CD38+ T cells. In contrast, short-term viremia had no effect on DC (Fig. 1B), NK, or activated HLA-DR+ NK cells (Fig. 1C), and retention of NK cytotoxicity (Fig. 1D) and flu-stimulated IFN-α production (Fig. 1E) was observed irrespective of viral rebound during TI periods or following suppression. Finally, no association between DC or NK cell numbers or function at baseline of each TI and resulting viremia was observed.
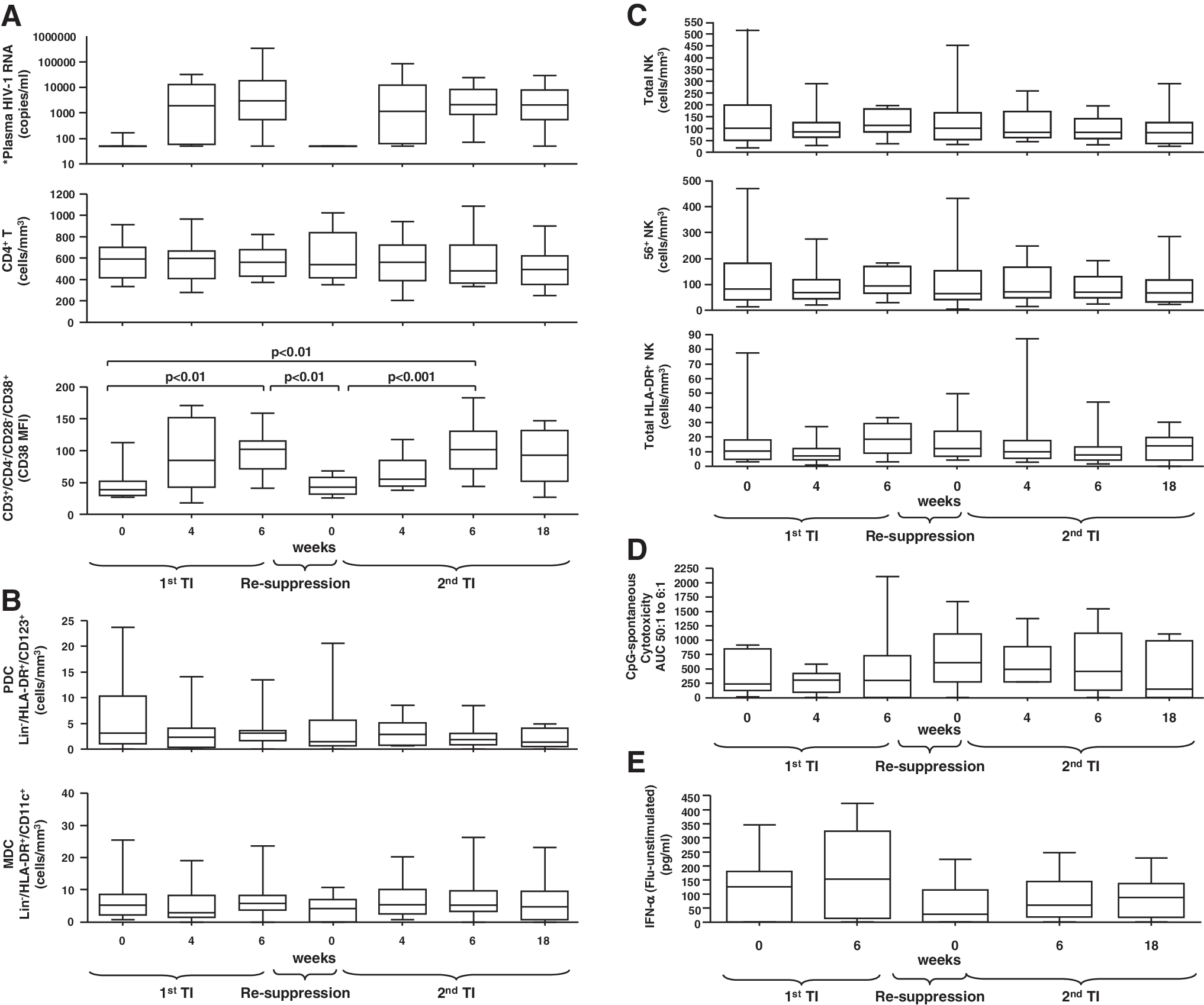
Retention of innate effectors cell numbers and function during subsequent therapy interruptions and in spite of acute viral replication. (
Overall, our data show that acute viral replication during TIs of up to 18 weeks is not associated with acute changes in the number of innate effector cell subsets or the onset of immediate impairments of NK and DC cell-mediated responses. Our data support our and other groups' observations showing lack of an immediate immunological “cost” of short-term TIs, as exemplified by retention of recall responses 9 –14 and control of LPS levels during short-term TIs. 15 As a result, short-term viremia is unlikely to acutely deplete the innate effector cell subsets at the same rate it affects changes in distribution and activation within the T cell compartment. Although delays in the levels of increase of innate cell immune reconstitution as compared to uninfected subjects have been described after ART initiation and may vary between ART-treated subjects, 1,2,5 our data suggest that short-term viremia is unlikely to result in a sustained innate change. Therefore, a period longer than 18 weeks of viremia is likely required to significantly erode realized gains from previous innate cell immune reconstitution levels. Importantly, our data support the hypothesis that innate function can contribute to immune control mechanisms (i.e., opportunistic infections) in spite of short-term viremia, while also serving as a potential target for immunotherapy, which may boost innate mechanisms of viral control. As a result, therapeutic trials targeting an activation of antiviral innate and/or adaptive responses (e.g., five such trials are currently recruiting as listed in clinicatrials.gov) on therapy interruption would not be expected to be acutely countered by an impairment in innate function.
Footnotes
Acknowledgments
We thank the patients who participated in the study and their providers, Cecile Gallo for study assistance, as well as Jane Shull and the Board and Staff of Philadelphia FIGHT for providing patients samples. This work was primarily supported by grants to L.J. Montaner by the National Institute of Allergy and Infectious Disease (NIH AI48398 and U01AI065279). Additional support was provided by The Philadelphia Foundation (Robert I. Jacobs Fund), The Stengel-Miller family, AIDS funds from the Commonwealth of Pennsylvania and from the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health, as well as by a Cancer Center Grant (P30 CA10815).
Author Disclosure Statement
No competing financial interests exist.