
Abstract
Guangxi is an important transit area for HIV transmission in South China. Characterization of the full-length genome of HIV-1 prevalent in the area is important for phylogenetic analysis and vaccine development. CRF01_AE is one of the most rapidly spreading subtypes in Guangxi. In this study, we reported thirteen near full-length CRF01_AE genomes from Guangxi, China. The nearly full-length genome was reverse transcripted and amplified in two halves with the 1-kb overlap regions. The PCR products were sequenced directly. The sequence analysis showed that all of 13 strains were CRF01_AE recombinant subtypes. Two clusters were set up with all of the sequences that grouped separately with sequences from Vietnam and Fujian, China, which strongly suggested multiple introductions of CRF01_AE strains into Guangxi province. The results will improve our understanding on the phylogenetic relationship of CRF01_AE strains in South China and also help in the development of a successful HIV vaccine.
I
CRF01_AE, along with CRF08_BC, is the dominant strain in Guangxi province. 3 However, most of the genetic analyses of CRF01_AE in Guangxi were based on partial genome sequences, which may not provide comprehensive genetic information on the entire viral genome. The only two CRF01_AE full-length genomic sequences of virus reported in 2000 were isolated in 1997, which may be far different from the strains prevalent now. 4 In this article, we describe 13 near full-length HIV-1 CRF01_AE sequences derived from plasma samples collected in 2005 and 2006 in Guangxi, China. Those sequences will contribute to molecular epidemiology studies and will be used for the development of an HIV-1 vaccine.
All of the 13 samples were collected between 2005 and 2006, with informed consent, from patients residing in Guangxi, China who had previously been diagnosed as being HIV-1 infected by antibody detection,. The epidemiological data are listed in Table 1. RNA was extracted from 500 μl of HIV-1-positive plasma specimens after being concentrated by centrifugation at 23,000 × g for 1 h using a high pure viral RNA kit (Roche, USA). RNA was reverse-transcripted into cDNA and the near full-length genome was amplified in two halves with 1-kb overlap regions. In brief, Superscript III (Invitrogen, USA) was used for reverse transcription with primer 1.R3.B3R (5′-ACTACTTGAAGCACTCAAGGCAAGCTTTATTG-3′) and vpu4 (5′- TTAATTTTACACATGGCTTTAGRCTTT-3′) separately for the 3′ and 5′ halves. Nested polymerase chain reaction (PCR) was performed by using High Fidelity Taq (Invitrogen, USA) according to the manufacturer's instructions with primers separately for the 3′ and 5′ halves. For the 5′ half, MSF12b (5′-AAATCTCTAGCAGTGGCGCCCGAACAG-3′) and vif4 (5′- TTGCCACTGTCTTCTGCTCTTTC-3′) were used for first round amplification and F2NST (5′-GCGGAGGCTAGAAGGAGAGAGATGG-3′) and vif3 (5′- TCGCTGTCTTCGCTTCTTCCTGCCAT-3′) were used for second round amplification. For the 3′ half, 07For7 (5′-CAAATTAYAAAAATTCAAAATTTTCGGGTTTATTACAG-3′) and 2.R3.B6R (5′-TGAAGCACTCAAGGCAAGCTTTATTGAGGC-3′) were used for first round amplification and VIF1 (5′-GGGTTTATTACAGGGACAGCAGAG-3′) and Low2c (5′-TGAGGCTTAAGCAGTGGGTTCC-3′) were used for second round amplification. The same amplification conditions were used in the two rounds of amplication of the two halves as follows: 94°C for 2 min and then three cycles (94°C for 30 s, 60°C for 1 min, 68°C for 5 min 30 s), then 32 cycles (94°C for 15 s, 60°C for 30 s, and 68°C for 5 min), followed by 68°C for 10 min. All amplifications were set up in a clean room using dedicated supplies and pipettes only. The positive PCR products were sequenced by Huada genomics company (China) with a variety of internal specific primers (available on request) after being purified.
IDU, injecting drug user.
All of the two overlapped subgenomic DNA fragments were successfully obtained from 13 samples. All of the sequenced fragments were edited and assembled into contiguous sequences on a minimum overlap of 30 bp with a 99–100% minimal mismatch with ContigExpress software, which is a component of Vector NTI Suite 6.0. Thirteen near full-length genomes were obtained. BLAST search against the HIV-1 sequence database and among themselves wasused to check for contamination
5
and no evidence of sample contamination was observed. Analysis of sequences showed that all of the gene structures of those NFLG sequences were normal with the nine open reading frames (ORFs) intact and opened except 05GX136. Many G–A mutations in 05GX136 strains, which formed stop codons in many genes, suggested the existence of hypermutation. This was proved by the Hypermut program (
To determine the subtype, all 13 sequences were aligned with the reference sequences representing subtypes A–D, F–H, J, K, and CRF01_AE (
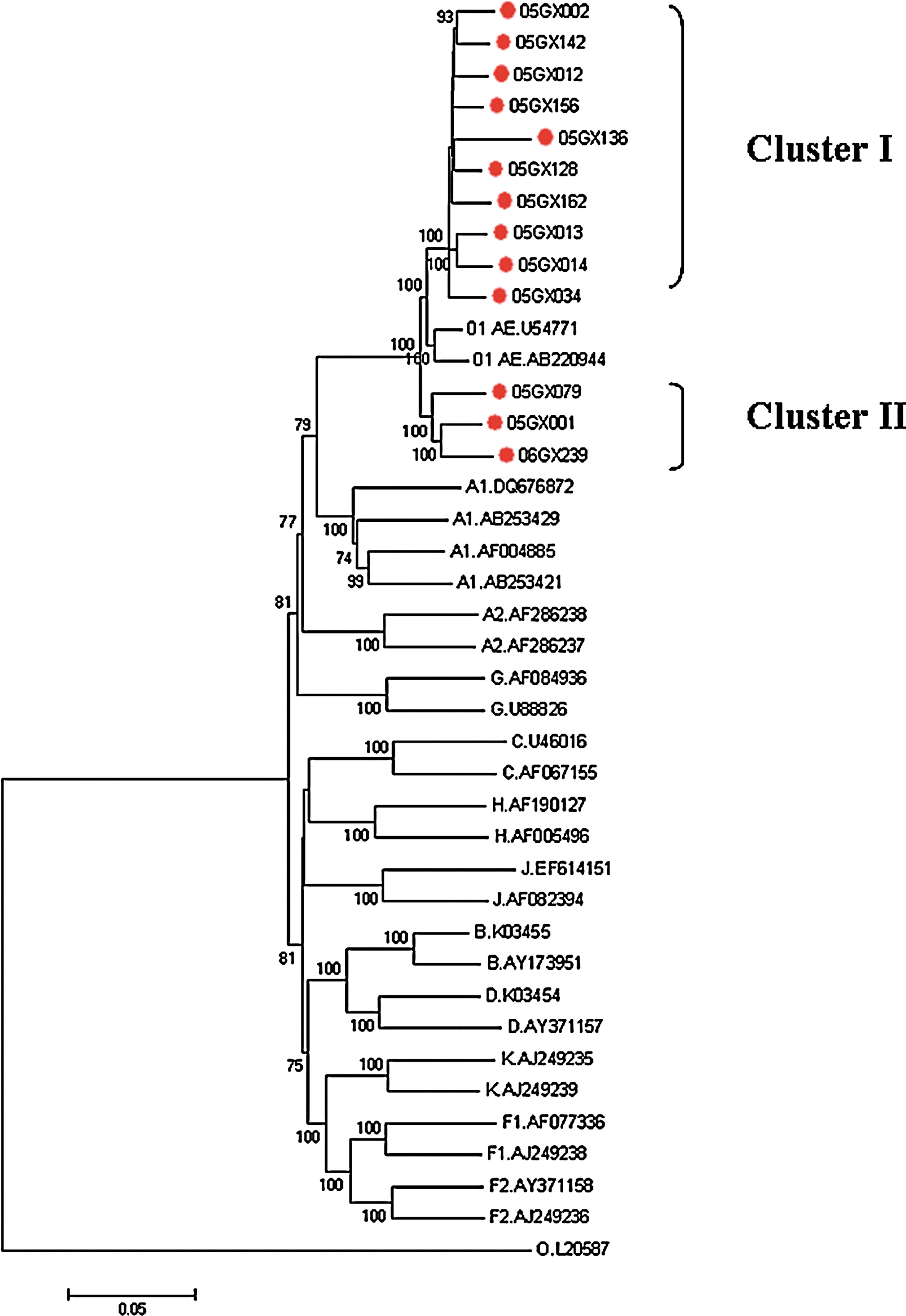
Phylogenetic tree analysis. A neighbor-joining tree was created with the 13 near full-length HIV-1 sequences from Guangxi (marked with gray circles) and the reference sequences of subtypes A–D, F–H, J, K, CRF01_AE, and group O (

Phylogenetic tree of HIV-1 subtype CRF01_AE based on the near full-length genome using the neighbor-joining approach. Previously published near full-length genomic sequences representing subtype CRF01_AE from several countries or areas, labeled with subtype, source, and name, were obtained from the database (
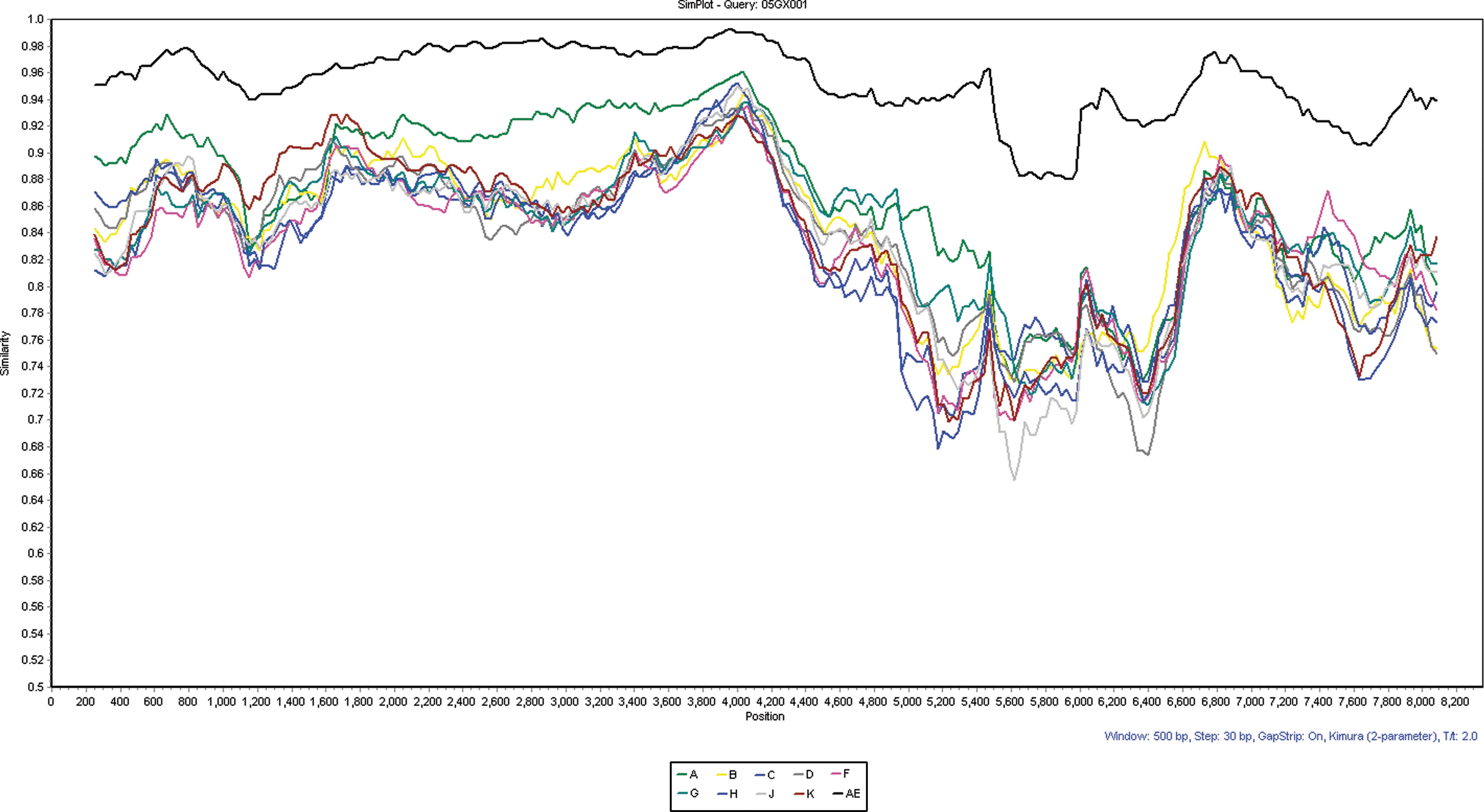
Similarity plots of the 05GX001 strain against subtype reference strains from the Los Alamos database, including A (92UG037), B (HXB2), C (95IN21068), D (ELI), F (93BR020), G (92NG083), H (056), J (SE7887), K(MP535), and CRF01_AE (CM240). Simplot was performed using a sliding window of 500 bp and a step size of 30 bp with replicates of the dataset used. The alignment was gap-stripped before being analyzed. (Color images available online at
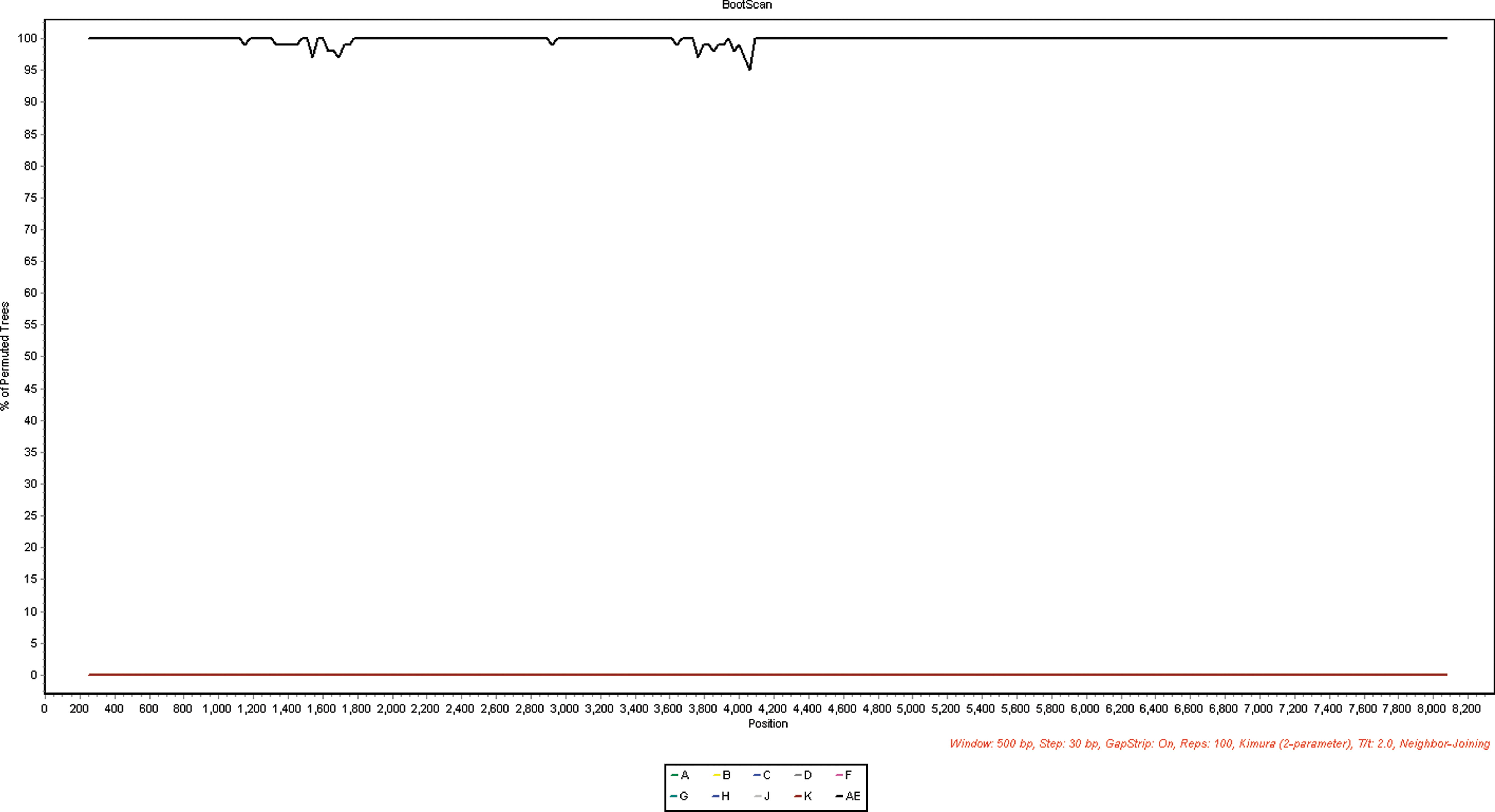
Bootscan analysis of the 05GX001 strain. The bootscan analysis was done using reference sequences of subtype A (92UG037), B (HXB2), C (95IN21068), D (ELI), F (93BR020), G (92NG083), H (056), J (SE7887), K(MP535), and CRF01_AE (CM240). The bootscan window was 500 bp with a step size of 30 bp. The x-axis indicates alignment; the y-axis indicates the percentage supporting the clustering with reference sequences. (Color images available online at
To further investigate the genetic relationships between our strains and those from other countries or areas, we performed additional phylogenetic investigations that included all our strains aligned with subtype CRF01_AE full-length genomes of different countries or areas gained from the Los Alamos database. The tree confirmed the two distinct cluster definitions. A close relationship between cluster 1 strains and some strains in Fujian province and Japan was implied, suggesting their similarity to each other. Cluster 2, together with two variants isolated in 1997 in Guangxi, grouped with some strains prevalent in Vietnam (Fig. 3), which proved the close epidemiological relationship of the CRF01_AE strains in Guangxi and Vietnam as reported previously. 6 –9
Fujian is another province in China with CRF01_AE as the main prevalent subtype. To now, 13 near full-length genomes of variants in Fujian have been reported in the Los Alamos database. 10 Unlike strains in Guangxi where two obvious subgroups were found, CRF01_AE sequences from Fujian dispersed among CRF01_AE strains from different countries in a phylogenetic tree, which suggested a greater complexity of HIV-1 CRF01_AE strains prevalent in Fujian than in Guangxi. The intersequence nucleotide distances of our sequences were computed using MEGA3.1 software and compared with CRF01_AE reference strains from Fujian Province, China. A low degree of interisolate diversity was found among our 13 strains with a mean overall distance of 4.5% ± 0.1%, whereas a high degree of interisolate diversity was found in Fujian with an overall distance of 7.2% ± 0.2%.
CRF01_AE was firstly identified in Thailand in the Southeast Asia region, 11,12 and caused a serious AIDS problem and an explosive HIV-1 epidemic. In China, the first subtype CRF01_AE was reported in Yunnan in late 1994. 13 In Guangxi, CRF01_AE was believed to be imported from Vietnam through Pingxiang City in the south of Guangxi following the heroin traffic route. 6 –9 In this study, we characterized the entire genomic structure of 13 HIV-1 strains spreading throughout Guangxi and observed two clusters in CRF01_AE strains prevalent in Guangxi, which may suggest the possibility of more than one introduction of the HIV-1 CRF01_AE strain into the region. The characterization of the near full-length genome will contribute to our understanding of the evolution of the CRF01_AE epidemic in Guangxi and will also be helpful for the development of an effective vaccine.
Sequence Data
The nucleotide sequences have been submitted to GenBank and assigned accession numbers GQ845124–GQ845126 and GU564221–GU564230.
Footnotes
Acknowledgments
This work was supported by the National Key S&T Special Projects on Major Infectious Diseases (Grants 2008ZX10001-004 and 2008ZX10001-012) and the National Natural Science Foundation of China (Grant 30700706). We also want to express our thanks to Dr. Feng Gao from the Duke Human Vaccine Institute, Duke University Medical Center for providing the technical support for reverse transcription and nest ed PCR.
Author Disclosure Statement
No competing financial interests exist.