
Abstract
Kenya is one of the sub-Saharan African countries affected by HIV-1 infection and AIDS. We investigated HIV-1 genetic diversity in 130 individuals from Busia, Bungoma, and Kakamega in western Kenya as part of an HIV-1 vaccine feasibility study in preparation for Phase III efficacy clinical trials. After RNA extraction the partial gag (484 bp) and env (1297 bp) regions were amplified and directly sequenced. Phylogenetic analysis was done using MEGA version 4 and recombinants were identified using the jpHMM tool and phylogenetic analysis. HIV-1 sequences were amplified from 122 of the 130 samples, 118 (90.8%) from the gag region and 78 (60 %) from the env region and 74 samples (56.9%) from both the gag and env regions. Of these sequenced on both regions, 51.4% were subtype A, 9.4% subtype D, 1.4% subtype C, 4.1% subtype G, and 33.7% were discordant and thus possible recombinants, including A1/C, A1/D, A1/A2, and A2/C. The jpHMM tool indicated a further two samples with CD and BD breakpoints within the env gene and one within the gag gene (A1C). An additional sample had an A1D breakpoint in the gag gene, but the envelope was not amplified. HIV-1 subtype diversity in western Kenya should be considered in vaccines designed for clinical trials in this region and this genetic diversity should be continuously monitored.
Introduction
S
Analysis of the HIV-1 env genes of virus strains from different geographic regions reveals that HIV-1 can be divided into three main groups: M (major), O (outlier), and N (non-M, non-O). HIV-1 group M has been further subdivided into genetically equidistant clusters of HIV-1 env genes, comprising subtypes A–D, F–H, J, K, and at least 45 circulating recombinant forms (CRFs) and numerous unique recombinant forms (URFs),
Previous molecular epidemiological studies done in Kenya mostly used samples from Nairobi. 4 –9 A few other studies also investigated southern, 10 northern, 11 and western Kenya, 12 as well as the coastal region. 13,14 All of these studies indicated that HIV-1 subtype A is the most common subtype in Kenya, but that subtypes C, D, G, and recombinant forms were also detected. 4 –14
The complexity of HIV-1 diversity creates major challenges for vaccine design and development strategies. More than one HIV-1 vaccine candidate based on HIV-1 subtype A has been evaluated in Kenya during Phase I and II clinical trials. 15 The western Kenya region, which is targeted for HIV-1 vaccine clinical trials, is close to Uganda, a neighboring country that is dominated by HIV-1 subtypes A and D in almost equal proportions. 16 Our aim was thus to investigate HIV-1 genetic diversity by amplification and sequencing of partial gag and env genes from 130 HIV-1-positive individuals from different hospitals in western Kenya. This project was part of an HIV-1 vaccine feasibility study in preparation for Phase III efficacy clinical trials.
Materials and Methods
Study population and sample collection
Volunteers were recruited from the general population from January to August 2007. They were seeking Voluntary Counseling and Testing (VCT) services in the Busia and Bungoma District hospitals and the Kakamega Provincial Hospital in the western part of Kenya. Following the signing of an informed consent form and counseling, all eligible participants were interviewed according to a standardized questionnaire developed for this study. The questionnaire included demographic characteristics such as age, gender, marital status, nationality, sexual behavior, and occupation. The study was approved by the Kenyatta National Hospital and Research and Ethics Committee (KNHREC).
Eight milliliters of venous blood was collected in EDTA tubes and centrifuged to separate the cells and plasma, which were stored in separate vials. The first plasma aliquot was used to confirm HIV-1 infection using the Vironostika Uniform II kit (Organon Teknika, Boxtel, The Netherlands) according to the manufacturer's instructions. For molecular characterization, the other plasma aliquot was frozen at −70°C until used.
PCR amplification and sequencing of the partial gag and env genes
HIV-1 RNA was extracted from the plasma using the QIAamp Viral RNA kit (Qiagen GmbH, Hilden, Germany) according to manufacturer's instructions and stored at −70°C. Proviral DNA from the buffy coat of two samples was extracted using the QIAamp DNA Blood Mini kit (Qiagen, GmbH, Hilden, Germany) according to the manufacturer's instructions.
Reverse transcriptase polymerase chain reaction (RT-PCR) amplification was performed on a 484-bp fragment of the gag gene (HXB2 nucleotides 1237–1721) using the Access-RT kit (Promega, Madison, WI) and methods previously described. 17
PCR was also done for a 1372-bp env region (HXB2 nucleotides 7002–8374) that included the gp120 V3 region up to the gp41 immunodominant region. For prenested RT-PCR, the primers ED5 (5'-ATGGGATCAAAGCCTAAAGCCATGTG-3') 18 and gp41R1 19 were used. Briefly, we used the Access RT-PCR kit (Promega, Madison, WI) with 5 μl of RNA, 200 μM of each nucleotide, 40 μmol of each primer, 1 mM MgSO4, 5 U each of AMV RT and Tfl DNA polymerase, and AMV/Tfl buffer in a total volume of 50 μl. After reverse transcription of 45 min at 48°C, the reaction was held at 94°C for 2 min, followed by 40 cycles of denaturing the DNA (94°C; 30 s), annealing of primers (58°C; 30 s), and extension of the annealed primers (68°C; 2 min). This was followed by a final extension step of 7 min at 68°C and the PCR product was kept at 4°C. A nested PCR of 1297 bp (HXB2 nucleotides 7002–8299) was done with a Promega GoTaq Flexi kit (Promega, Madison, WI). Template DNA for the nested PCR consisted of 3 μl of reaction product from the first round with 200 μM of each nucleotide, 40 μmol of each primer, ES7 20 and Menv19R, 17 1.5 mM MgCl2, 2.5 U of Taq polymerase, and GoTaq buffer made up in a total volume of 50 μl. The PCR cycle method used was similar to that of prenested PCR, except for the primer annealing step at 44°C. The PCR products were visualized using agarose gel electrophoresis.
PCR products were purified using Exonuclease (Exol) and Shrimp alkaline phosphatase (SAP) (USB Corporation, Cleveland, OH) at 37°C for 15 min followed by heat inactivation of the enzymes at 80°C for 15 min. These purified products were kept at −20°C until sequencing reactions were done. All PCR products were sequenced on both strands using the BigDye Terminator V3.1 Cycle Sequencing kit and analyzed on an ABI Prism 3130xl automated DNA sequencer (Applied Biosystems, Foster City, CA).
Sequence and phylogenetic analysis
Sequences were analyzed and the overlapping DNA fragments were assembled using Sequencher version 4.8 (Gene Codes Corporation, Ann Arbor, MI). Nucleotide sequences were translated into amino acid sequences and submitted to GenBank using Sequin v7.70 (
Results
Patient demographics and clinical and epidemiological features
The study group consisted of 130 volunteers and included 26% males (n = 34) and 74% females (n = 96). The mean age of the males was 36 years and that of the females was 32 years. None of the volunteers included in the study received antiretroviral therapy. The following numbering system was used: 2201, 3301, 4401, where “22” indicates samples from Busia, “33” indicating samples from Bungoma, and “44” indicates samples from Kakamega. Both Kakamega and Busia lie along a major highway to Uganda and all study participants were of Kenyan nationality, except for samples 2202, 2228, and 4436, which were of Ugandan nationality (Table 1).
PCR amplification and sequencing
HIV-1 sequences were amplified and 118 (90.8%) were positive in the gag region and 78 (60 %) were positive in the env region. Only eight (6.2%) of the samples were negative with both gag and env PCR. Four samples were gag PCR negative, but env PCR positive and 44 samples were negative for env PCR but positive for gag PCR. The gag region is more conserved than the env region and genetic variation might explain why only 60% of the samples could be amplified with the env PCR primer set. In summary, from 130 samples only 74 (56.9%) were successfully sequenced and characterized on both the gag and env regions. Of these 74 samples, 38 (51.4%) sequences were subtype A, seven (9.4%) sequences were subtype D, one (1.4%) sequence was subtype C, three (4.1%) sequences were subtype G, and 25 (33.7%) sequences were discordant and thus possible recombinants. A summary of all the identified subtypes is indicated in Table 2.
Phylogenetic analysis and subtyping the gag gene
The result of the phylogenetic analysis of the partial gag gene is shown in Fig 1. The sequences did not cluster by collection site but were intermixed throughout the tree, indicating multiple introductions into western Kenya. The majority of the samples clustered with HIV-1 subtype A1. Two sequences (samples 2203 and 3317) clustered with subtype A2, three sequences clustered with subtype G (samples 2220, 2222, and 3324), and five sequences (samples 2215, 3302, 3309, 4420, and 4425) clustered with subtype C. The sequence of sample 4435 was too short to include in gag phylogenetic analysis, but with blast analysis and the jpHMM HIV tool it was subtyped as C. Nineteen samples were grouped with subtype D. The sequences from samples 2207, 2217, 3325, and 4408 were outliers in the phylogenetic tree and did not cluster with any subtype, but the jpHMM tool indicated that these sequences were subtype D. Although discrepancies between online tools for assigning subtyping/recombinant forms were observed, this could mostly be resolved. The jpHMM analysis was used to indicate the breakpoints of the recombinants and these partial sequences were then used for phylogenetic analysis.
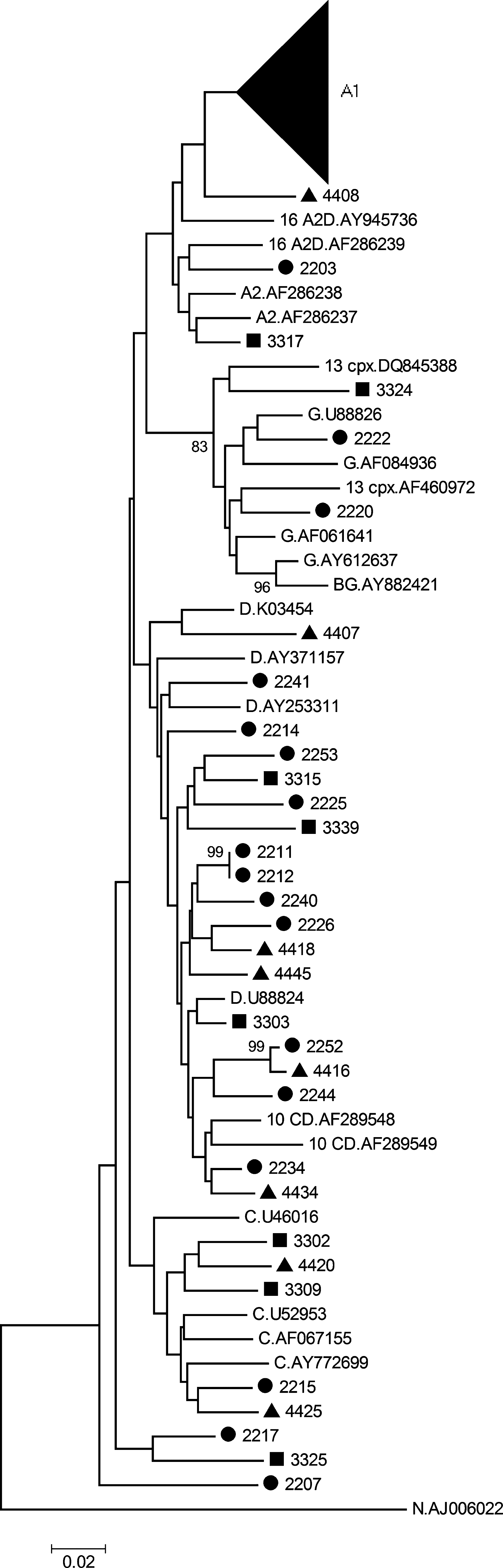
Phylogenetic tree of the HIV-1 gag region spanning nucleotides 1237–1721 (HXB2 coordinates). Patient samples from Busia (•), Bungoma (▪), and Kakamega (▴) in Kenya were aligned and compared with reference sequences from the Los Alamos HIV database using Clustal X version 2.0 21 and manually checked and edited with Geneious Pro version 4.8.3. 22 Reference sequences are indicated by the subtype and GenBank accession number. The neighbor-joining tree was constructed using the Kimura two-parameter algorithm 24 in MEGA version 4. 23 The bootstrap values of 1000 replicates above 70% are indicated next to the node and the scale bar at the bottom represents the number of base substitutions per site. All positions containing gaps and missing data were eliminated from the dataset.
Final analysis of the 118 gag sequences indicated that 82 (69.5%) were subtype A1, two (1.7%) were subtype A2, six (5.1%) were subtype C, 23 (19.5%) were subtype D, 3 (2.5%) were subtype G, and two (1.7%) were possible recombinants (Table 1). These possible gag recombinant sequences from samples 2213 and 2219 need to be further analyzed. Phylogenetic analysis of the gag gene also indicated that sequences from samples 2211 and 2212 clustered closely together with a bootstrap value of 98%. These samples were epidemiologically linked.
Phylogenetic analysis and subtyping the env gene
The complete env gene PCR fragment (1297 bp) could not be sequenced for all the samples. Some of the sequence chromatograms were difficult to read due to multiple peaks, specifically in the gp120 variable regions. Thus we had env sequences of almost 1297 bp (n = 52) and shorter sequences of about 600 bp (n = 26). The results of the phylogenetic analysis of the partial env gene are shown in Fig. 2.

Phylogenetic tree of the HIV-1
In these env phylogenetic trees, the majority of the sequences clustered with subtype A1. Seven sequences (samples 2224, 2238, 2254, 3308, 3317, 4420, and 4436) clustered with subtype C, three sequences (samples 2220, 2222, and 3324) clustered with subtype G, and sequences from 15 samples clustered with subtype D. The sequence from sample 2241 (Fig. 2a) was typed as A1, but had a long branch. The jpHMM tool indicated that this sequence is an A1D recombinant (Table 2) and this was confirmed with phylogenetic analysis of the subfragments. Another sequence (sample 3331) clustered with subtype A2 (Fig. 2b), with a very long branch and the jpHMM tool also indicated recombination in this sequence. Final analysis of the 78 env sequences indicated that 51 (65.4%) sequences were subtype A1, five sequences (6.4%) were subtype C, 12 sequences (15.4%) were subtype D, three sequences (3.8%) were subtype G, and seven sequences (9.0%) were recombinants (Tables 1 and 2).
Recombinant analysis using jpHMM
Recombinants were further evaluated with the jpHMM-HIV tool, a probabilistic generalization of the jumping-alignment approach. Because recombination breakpoints identified by jpHMM were found to be significantly more accurate than breakpoints defined by traditional methods based on comparing single representative sequences, 25,26 we used this approach to identify recombinants. Using this tool and phylogenetic analysis (data not shown), we identified nine unique recombinants (Table 3) with breakpoints within the env and gag genes. Sequences 2219, 3308, and 4436 were A1C recombinants, 2213 and 2241 were A1D recombinants, 3328 was a DG recombinant, 4418 was a CD recombinant, 3331 was an A1A2 recombinant, and 4407 was a BD recombinant. Bootstrap support for the BD recombinant was low.
Numbering according to reference strain HXB2.
Phylogenetic and recombinant analysis indicated that the 74 gag/env subtypes included 38 (51.4%) A1/A1, seven (9.4%) D/D, three (4.1%) G/G, four (5.4%) A1/C, six (8.1%) A1/D, three (4.1%) C/A1, seven (9.5%) D/A1, and one (1.4%) each of C/C, A1/A2, and A2/C. The jpHMM tool indicated a further two samples with breakpoints within the env gene (4418 and 4407) and another one within the gag gene (2219). An additional sample, 2213, had a breakpoint in the gag gene, but the envelope was not amplified (Table 3).
Discussion
We investigated HIV-1 genetic diversity in 130 HIV-1-positive individuals from western Kenya in preparation for Phase III vaccine trials. In this study we detected subtypes A (A1 and A2), C, D, G, and recombinants, including recombinants with breakpoints in the gag (n = 2) and env (n = 7) regions. The results found in this study are consistent with the previous studies.
Kenya is bordered by five countries (Tanzania, Uganda, Sudan, Ethiopia, and Somalia) and the distribution of HIV-1 subtypes in these countries is variable. For instance, subtype C and AC recombinants dominate in Somalia and Ethiopia,
27
subtypes C and D are common in Sudan,
28
and subtypes A and D are common in Uganda.
16
In Tanzania the diversity includes subtypes A, C, and D, as well as AC, AD, and CRF10_CD recombinants
Although subtype A is the most prevalent subtype in Kenya, the HIV-1 subtype distribution can vary geographically. In Nairobi, Nielson and co-workers 5 detected subtypes A (70.3%), D (20.5%), C (6,9%), G (0.3%), and recombinants (2.2%) by analyzing the partial env gene of 320 samples. The Pumvani MTCT cohort in Nairobi was divided into subtypes A (58%), D (20%), and C (1%) by analyzing the partial gag and protease genes of 130 samples. 6 Analyzing the integrase gene of 140 samples from Nairobi, Lihana and co-workers 7 detected subtypes A (64%), D (17%), C (9%), and G (1%). By analyzing the complete genome in 10 samples from Nairobi, 50% of the samples were subtype A and 50% were recombinants. 29
In the southern part of Kenya, 56% of samples analyzed were subtype A and 40% were recombinants when the complete HIV-1 genome of 41 samples was sequenced. 10 In northern Kenya the partial env region of 72 samples was analyzed and subtypes A (50%), C (39%), and D (11%) were detected. This region borders Ethiopia, which is dominated mainly by subtype C, and this study indicated that cross-border movement can influence the circulation of HIV-1 subtypes. 11 In a previous study done in the western part of Kenya on the partial env region of 30 samples, it was revealed that subtype A was the most prevalent (67%) strain, followed by subtypes D, C, and G. Twenty-three percent of the samples were recombinants (AD, AC, and CRF10_AD) and unclassified strains, indicating that western Kenya may be a hotspot for recombination. 30 A larger investigation of 460 samples from Kisumu in western Kenya sequenced partial gag and env regions and 344 samples (75%) were concordant in both regions (subtypes A, 59%; D, 10%; C, 2%; G, 3%) and 25% were discordant, indicating D/A (40, 8.7%), A/D (27, 5.9%), C/A (11, 2.4%), and A/C (8, 1.7%) recombinants. 12 Though all these molecular studies have been done in the same region, the data indicate that there are significant differences in the distribution of HIV pure subtypes and recombinant forms. However, most of the studies have obtained samples from women participants. These data may have been different if there was equal participation of both male and female participants.
In this study we sequenced 74 samples in both the gag and env regions. Concordant results indicated that subtype A was dominant (51.4%), followed by D (9.4%), C (1.4%), and G (4.1%). Twenty-two samples were discordant in the gag and env regions, indicating possible A1/C (n = 4), A1/D (n = 6), C/A1 (n = 3), D/A1 (n = 7), A1/A2 (n = 1), and A2/C (n = 1) recombinants. A further seven samples had breakpoints in the env region and another two in the gag region. Detection of recombination in 25 (33.7%) of the samples might underestimate recombination. The recombinants with breakpoints in the gag region (strain 2213, gag A1D and 2219, gag A1C) were both from Busia. The env sequence from 2219 was a subtype A1 and in 2213 we were unable to amplify the env region. The samples with breakpoints in the env region were A1C (n = 2), and one each of A1/A2, A1D, DG, and DC.
Our study has shortcomings as well. We never confirmed our results with full-length sequencing, although similar studies confirmed a good correlation between sequence results and partial gene sequencing. Evidence of intersubtype genes on a single region as described above suggests that full-length sequencing may be a more inclusive technique to map out all recombinations and mutations and therefore offers a true picture of viral diversity, especially in Africa where many subtypes coexist. 31
The genetic variability of HIV globally presents a major challenge for vaccine developers because immune responses that recognize HIV from one subtype may fail to recognize viruses from other subtypes. Previous HIV vaccine candidates that have been evaluated in Kenya were based on the HIV-1 subtype A, the dominant strain. 4,5,30 Two HIV vaccine candidates were developed in a partnership between the University of Nairobi's Kenya AIDS Vaccine Initiative (KAVI), the Medical Research Council, University of Oxford, and the International AIDS Vaccine Initiative (IAVI). With the evidence of superinfection and recombination, 32,33 a vaccine designed based on the dominant subtype may not offer protection against another subtype. HIV-1 subtype diversity in western Kenya should be considered in vaccines designed for clinical trials in this region and this genetic diversity should be continuously monitored.
The study demonstrates that four HIV-1 pure subtypes (A, C, D, and G) and a high proportion of recombinants of these subtypes are present in western Kenya. These data therefore suggest that a multiclade HIV-1 vaccine with antigenic determinants from all subtypes present in the region may be the best vaccine for future clinical trials in western Kenya. More studies are needed to monitor the molecular evolution of recombinants and the introduction of new viral strains ahead of efficacy clinical trials.
Sequence Data
The sequences were deposited in GenBank with Accession numbers FJ346340–FJ346535.
Footnotes
Acknowledgments
A part of the work has been partially presented at AIDS Vaccine 2008, Cape Town, 13–16 October 2008 (AIDS Research and Human Retroviruses 2008; volume 24, Supplement 1).
Author Disclosure Statement
No competing financial interests exist.