
Abstract
Besides being extremely useful in measuring the level of HIV-1 diversity and prevalence in populations, the molecular analysis of genomic sequences provides crucial surveillance support and aids in the development of new therapies and effective vaccines. The present study focused on gag and env DNA and amino acid sequences that were generated from samples taken from 61 infected patients in the City of Salvador, Bahia, located in northeastern Brazil. In order to determine selective pressure and predict coreceptor usage, Bioinformatics tools were employed in phylogeny reconstruction. Fifty-six (91.8%) viruses were classified as belonging to subtype B, three (4.9%) from F1, and two (3.3%) from BF1 recombinants. Based on the characterization of the V3 region, the subtype B strains were represented by eight (18.2%) Brazilian variants (B’-GWGR), 20 (46.5%) European/EUA B variants (GPGR), and 15 (34.9%) GXGX variants. The mean time elapsed since diagnosis was 13 years among subtype B’ and 9 years in subtype B. The mean dN/dS ratios from the GWGR, GPGR, and GXGX groups, when compared to an HXB2 reference, were 0.72, 0.77, and 0.67, respectively. Seventy-six percent of the viruses studied were predicted to use the CCR5 coreceptor for cell entry (R5 viruses), while 24% were predicted to use the CXCR4 or were classified as dual tropic viruses. The prevalence of subtypes B' and recombinant B/F1 was shown to be lower than findings from previous studies performed both in Brazil (B’) and in Bahia (B/F1). The association between subtype B’ and a lengthy period of time since diagnosis can be correlated with a slower disease progression in infected patients, when compared with those infected with subtype B.
Introduction
T
Materials and Methods
Sixty-one HIV-infected individuals, patients from the Professor Edgar Santos University Hospital (HUPES), located in the City of Salvador, Brazil, were recruited in 2006. All patients signed a letter of informed consent. Two to 5 ml of whole blood samples were collected and transported to the Advanced Laboratory of Public Health (LASP) at CPqGM/FIOCRUZ where they were stored at −20°C until use. Clinical data were also obtained from patient medical records. This study was approved by the CPqGM/FIOCRUZ Ethics Committee. DNA samples were extracted from 200 μl of blood using a QIAamp DNA kit (Qiagen Inc., Valencia, CA) in accordance with manufacturer's directions. Fragments of gag (positions 898 to 1968) and env (positions 6945 to 8183) genes, both relative to the HXB2 reference sequence, were PCR amplified using nested primers. The amplified DNA was purified using a purification kit (Qiagen Inc.) and sequenced using a BigDye Terminator v.3.1 Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Carlsbad, CA) and an automated ABI 3100 Genetic Analyzer (Applied Biosystems). Primers GAG2, G17, H1G777, and MZ14 were used for gag sequencing reactions, while primers ED31, MM4, ES7, and ED14 were used for the env region.
Electropherograms were obtained, analyzed, and exported in FASTA format using SeqScape v2.1.1 software (Applied Biosystems). Sequences were subjected to the BLAST search algorithm (
Phylogenetic analyses were performed using PAUP* 4.0b10 software
8
to generate neighbor-joining (NJ) and maximum likelihood (ML) trees using the GTR model of nucleotide substitution.
9
Node reliability was assessed using bootstrap analysis (1000 replicates) and a likelihood-ratio test was used to calculate statistical support for tree branches. The trees were drawn using Figtree software (
Coreceptor usage was predicted in silico based on V3-sequences and clinical data using the geno2pheno [coreceptor] online tool (
Selective pressure was assessed in the 1155-bp envelope fragment using the SNAP
12
web-based tool (
Results
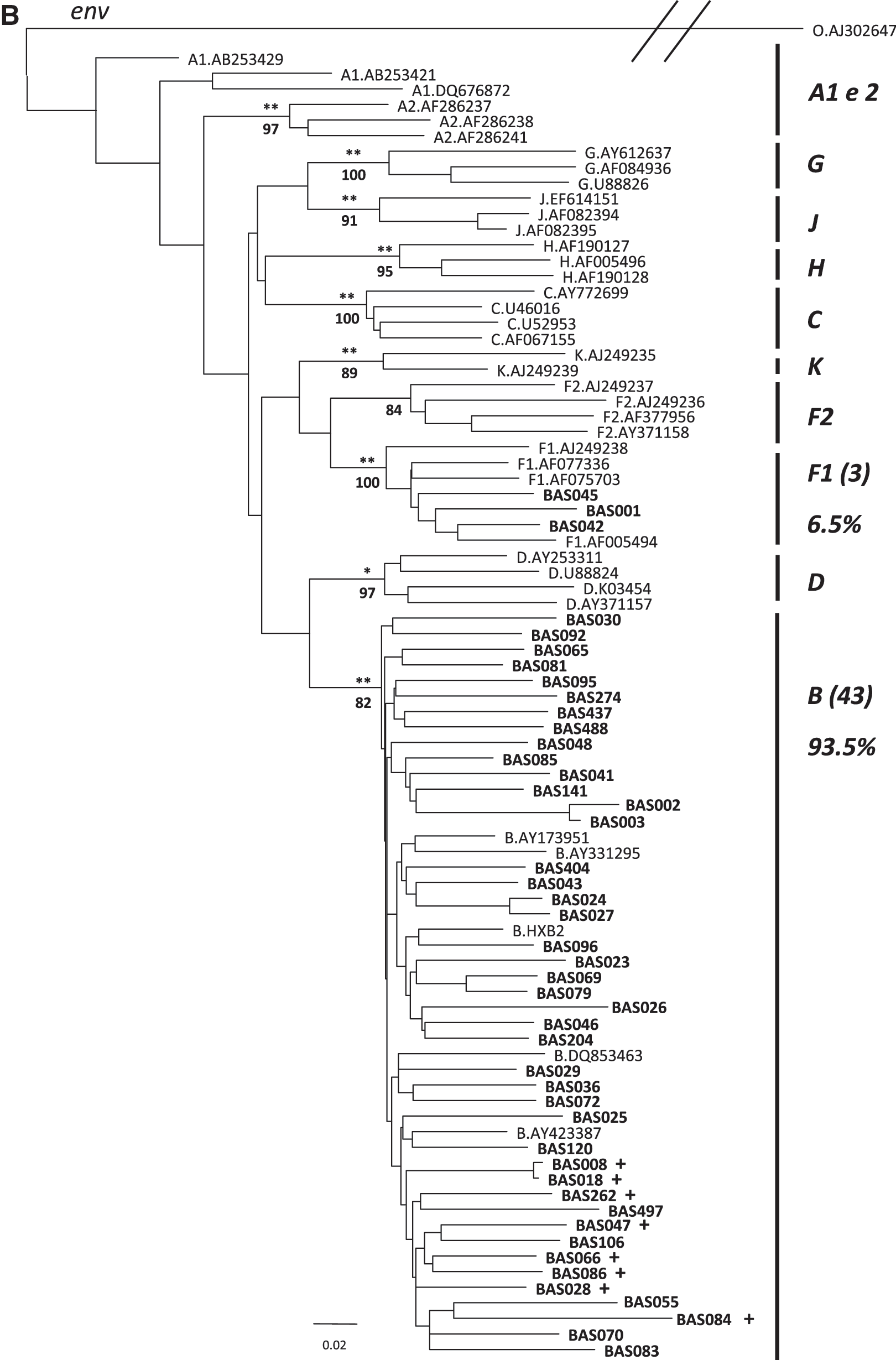
Out of 61 HIV-1 positive samples analyzed, 41 were obtained from males and 20 were from females (2:1 ratio). Among males, a mean age of 43.3 years was observed versus 39.1 years in females. Mean infection time was 9.94 years, mean viral load was 75,587.87 copies/ml and mean TCD4 cell counts were 421.61 cells/ml, while TCD8 cell counts were 1,262.24 cells/ml. Regarding ethnicity, 23% of the patients reported being of European or Latin descent, while 47.5% indicated Mixed-race and 29.5% African descent. No statistically significant differences regarding time of infection, viral load, or TCD4 and TCD8 cell counts were observed between ethnic groups. Out of 61 samples, 42 gag and 46 env gene sequences were amplified with 27 sequences amplified in both genes. Phylogenetic analysis of the gag gene showed 40 (95.2%) sequences clustered within the subtype B reference group, while two (4.8%) did not cluster within any pure subtypes and were further characterized as BF1 recombinants as revealed by recombination analysis. The BAS026 sequence showed two breakpoints (B/F1/B): one in positions 1357–1398 and another at positions 1482–1615 (relative to HXB2). The BAS096 sequence presents one breakpoint in positions 1187–1228 (relative to HXB2). In the env sequences, 43 (93.5%) were shown to cluster inside subtype B, and 3 (6.5%) inside F1 (Fig. 1). The two gag BF1 recombinants were subtyped as B in the env tree. With respect to the 27 samples that were characterized in both genes, 25 (92.6 %) belonged to subtype B, while only two (7.4 %) were found to be BF1/B. Considering all 61 samples, 56 (91.8%) were shown to be subtype B, three (4.9%) were F1, and two (3.3%) were BF1 recombinants. The 43 subtype B env sequences were translated into amino acid sequences. Based on V3 characterization, eight (18.2%) Brazilian (B’-GWGR), 20 (46.5%) European/EUA B (GPGR), and 15 (34.9%) GXGX variants were found. The mean pairwise distance was 0.11, 0.13, and 0.12 within the GWGR, GPGR, and GXGX groups, respectively. To test whether selective pressure was altered due to V3 loop substitution, the dN/dS ratio of each sequence was determined under comparison with an HXB2 reference sequence and the sequences were subsequently grouped in categories: GWGR, GPGR, and GXGX. The mean dN/dS ratio of all sequences was 0.725 when compared to HXB2. The mean GWGR, GPGR, and GXGX dN/dS group ratios were 0.72, 0.77, and 0.67, respectively, when compared to HXB2. Statistical testing revealed no statistically significant differences between the GWGR and GPGR groups (p > 0.05). A comparison between GPGR and GXGX group sequences showed a significant difference (p = 0.018), revealing a higher rate of positive selection in the GPGR group. We compared the clinical characteristics of individuals harboring B' (GWGR) and B (GPGR + GXGX) viruses. Table 1 shows that the mean time period since diagnosis was higher in the subtype B' group than in the subtype B group and that the mean age was also higher in the former group.

NJ tree based on gag (
Test t for equality of means.
With respect to coreceptor usage, eleven (24%) out of the 46 V3 sequences were predicted to use the CXCR4 coreceptor (X4 virus). The mean age among subjects infected with X4 virus (32.4 years) was lower than the mean age among subjects infected with R5 virus (43.3 years) (p < 0.05).
Discussion
Molecular studies of HIV-1 are critical to furthering the understanding of mechanisms involved in AIDS pathogeny and to support the development of vaccines and efficacious therapies. The number of HIV sequences from Northeastern Brazil remains scarce, as well as precise information associating HIV-1 diversity with clinical data. To this end, the authors studied samples and laboratory information from randomized patients in Salvador, the capital of the Northeastern Brazilian state of Bahia and the third most populous city in the country.
In the northeastern region of Brazil, as well as the country as a whole, HIV-1 subtype B remains predominant. However, in the past decade, several studies have reported on the increasing prevalence of other genotypes, notably BF recombinants. 14 In the City of Salvador, a lower prevalence of B/F1 recombinant forms (3.3%) was observed in our samples when compared with recent studies showing a 10% and 13% prevalence of BF forms in the Brazilian Northeast. 14,15 This may be explained by the fact that our sequencing involved two fragments within the gag and env genes, implying that other genomic regions where recombination could have occurred may have been overlooked.
Some of the sequences characterized in this study presented different clustering within the large subtype B group in gag and env trees. For instance, sequences BAS008 and BAS018 showed a close relationship in the env tree; however, their gag genes were not closely related in the phylogenetic analysis. These observations could be indicating that these samples derived from a common ancestral that went through different evolution process and subsequent intrasubtype recombination. Furthermore, these strains could be representing the occurrence of dual infection in one or both individuals. This study found the prevalence of subtype B’ to be much lower than 50%, which was observed in the country's southeastern region. 16 This leads us to suggest that Salvador has experienced different introduction(s) and founder effect(s) from the Brazilian Southeast. In addition, no phylogenetic distinctions were observed between subtypes B’ and B, findings consistent with previous reports. 16 However, this study demonstrated that mean age (49 years) and time period elapsed since diagnosis (13 years) in subtype B'-infected patients was higher than mean age (39 years) and time period elapsed since diagnosis (9 years) for subtype B-infected individuals. This could be related to an increased replication rate and accelerated disease progression characteristic of subtype B sequences. 5 In fact, the GPGR sequences exhibited a slightly higher mean pairwise distance which may explain the clinical difference between subtype B’ and subtype B.
The results presented in this study demonstrate a lower level of genetic diversity and, specifically, a lower prevalence of BF recombinant forms than those previously found in Bahia and in the Brazilian Northeast region. 14,15 This may be related to the presence of recombination points in genomic regions other than those analyzed by this study, and, therefore, further studies involving the full genome sequencing of HIV isolates from this geographic region could contribute to a better understanding of this region's HIV epidemiology.
Footnotes
Acknowledgments
The authors are grateful to the individuals who donated blood for the purposes of this study, to the FIOCRUZ-PDTIS sequence platforms, Mr. Augusto Santana and Mrs. Maurina Alcantara from HUPES for providing access to patient biomedical records, and to Mrs. Elisabeth Deliege Vasconcelos for editing and revising this manuscript.
Sequence Data
The new sequences in this study were reported to GenBank under the accession numbers GU595197-281 and GU722093-95.
Author Disclosure Statement
No competing financial interests exist.