
Abstract
We present an HIV-1-infected patient with a profile of transmitted drug resistance (RT M41L, E44D, V118I, L210W, T215D) sustained during more than 10 years in the absence of treatment. Clonal analysis of different plasma and cellular samples within this period did not reveal any reversion to the wild-type genotype.
T
The patient is a 33-year-old woman infected by sexual transmission and diagnosed in August 1997 (CD4 537 cells/μl, VL 25,000 cp/ml). Her clinical evolution is summarized in Fig. 1. A genotypic resistance test in naive patients was not included in the standard of care by 1997–1998 and according to the recommendations for ART at that time, she received treatment with AZT + 3TC + EFV from May 1998 until January 2002 and the plasmatic HIV-1 viral load (VL) was always below the limit of detection while on treatment. In 2002 she was recruited for a clinical trial designed to study the strategy of ART interruptions guided by CD4 cell counts and treatment was stopped; she underwent follow-up every 3–4 months. CD4 cells were maintained above 500/μl until September 2008 when the patient showed a CD4 cell count of 324/μl and a VL of 320,000 cp/ml. ART was then reconsidered and a genotypic test disclosed an HIV-1 subtype B and the presence of the following mutations in the retrotranscriptase (RT) gene: M41L, E44D, V118I, L210W, and T215D.
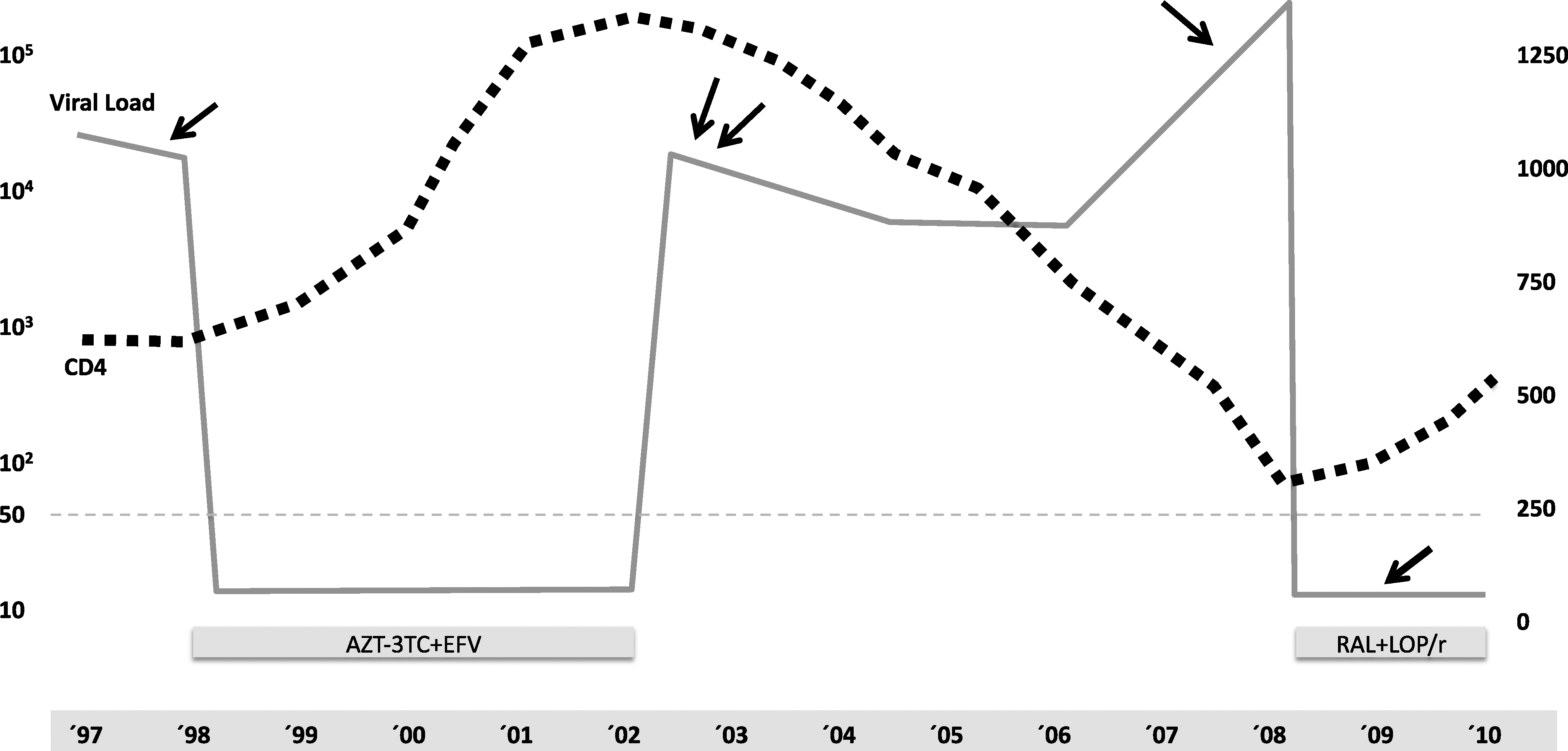
Clinical progression and ART treatment history of the case reported: viral load (continuous line) and CD4 (dotted line) evolution along the follow-up of the case. Arrows designate the sampling dates for clonal analysis of both plasma (´98, ´02, ´08) and proviral DNA (´09).
We performed a retrospective study of archived plasma samples from 1998 (before first treatment), 2002 (two samples, 1 and 3 months after stopping treatment), and 2008. The standard population-based genotypic assay (ViroSeq HIV-1 Genotyping System v2.0, Abbott Molecular) detected the same profile of mutations in all plasma samples from 1998 to 2008. To better characterize the evolution of the resistance sequence, we performed sequencing of individual clones as follows. Amplified fragments covering the first 330 amino acids of the RT region of each sample were ligated into the pDrive vector (Qiagen) and competent Escherichia coli bacteria were transformed. Inserts from single colonies were sequenced and analyzed by using an ABI PRISM 3100 genetic analyzer, VectorNTIv.11 software (ContigExpress and AlignX), and Mega 4.1 to evaluate the phylogeny and clone evolution. A total of 71 clones were analyzed (20 from 1998, 31 from 2002, and 20 from 2008).
The clonal analysis found the same resistance profile (M41L, E44D, V118I, L210W, and T215D) in all samples, with the exception of L210G, which was detected in eight clones in the samples from 1998 and 2002 and E44G in one clone from 2008 (Fig. 1). No reversion from any of these mutations to the susceptible genotype was detected in any clone during follow-up. Additionally, a single clone was detected with V75I in the first sample from 1998, a single clone with V106A in 2002 (immediately after first treatment), while Y188C was present in 1 out of 20 clones analyzed from the sample of 2008, more than 6 years after ART interruption (Fig. 1). A tropism test (Trofile, Monogram) performed in 2009, just before initiation of the second ART, identified an R5 tropic virus. The patient was finally treated with RAL + LOP/r and achieved complete virological suppression in 4 weeks.
HIV-1 proviral DNA, corresponding to an aviremic blood sample obtained 6 months after the new treatment initiation, was amplified by PCR and analyzed using the same procedures described for plasma samples. All 15 clonal sequences from proviral DNA were also shown to harbor the same profile of RAMs detected in population-based sequencing of plasma and neither reversion to a wild-type genotype nor additional RAMs were detected in the cellular archive (Table 1).
Within the RT fragment sequenced (1–330 aa), only residues with at least a resistance-associated mutation or two polymorphic changes as compared with the subtype B HXB2 consensus are shown. Mutations conferring a reduction of susceptibility to antiretroviral drugs according to the Stanford Database are in bold.
Taking into account the limit on sensitivity of clonal analysis, sequencing of 15–30 clones has a probability of 95% for detecting a mutant present in at least 23% and 13% of the clones, respectively. This patient maintained the same profile of RAMs for more than 10 years without any evidence of reversion to the sensitive wild-type genotype at any position in RT despite the fitness cost associated with some of these mutations, particularly M41L and L210W, which have shown in vitro a greater impact on replication capacity. 4 –6 This fact would be compatible with the recent observation of the monoclonal origin of the founder virus in most heterosexually transmitted infections. 7,8
In this patient a single resistant sequence could have been responsible for the initial infection and, since all the RAMs were contained in the same genome, reversion to the wild-type genotype could have been reduced. Not surprisingly, the genetic divergence (nucleotide substitutions per site) found among the sequences analyzed in each plasma sample was relatively low: 0.008 (1998), 0.011 (2002), 0.010 (2008), and 0.023 the divergence between sequences from 1998 to 2008 (Fig. 2). The main RAMs detected in this patient, M41L, L210W, and T215D, correspond to the evolutionary cluster of TAMs in the TAM-1 pattern 4 in witch T215D is most likely derived from the less fit and highly resistant T215Y. The lack of detection of T215Y in any circulating clone or the cellular-associated virus supports the fact that the primary infection of this patient could have been produced by a single clone in witch T215D would partially compensate the fitness cost of M41L and L210W. On the other hand, we detected a single clone with V75I (an additional NRTI-related mutant) in the first sample from 1998, suggesting that the initial inoculum could have been more complex or that V75I was the only RAM that was negatively selected during evolution due to a higher impact on fitness. 4
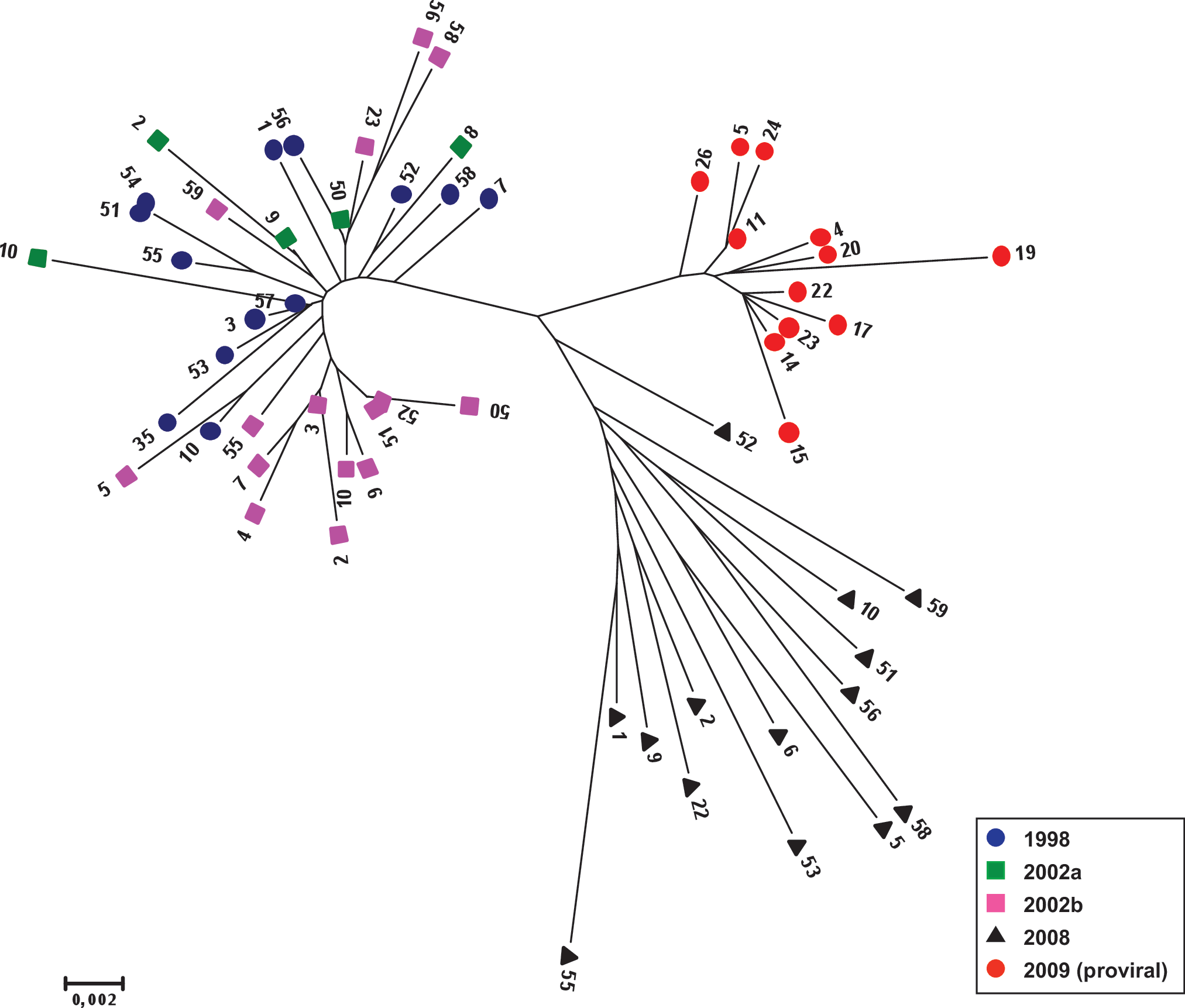
Phylogenetic tree describing the relationship among the reverse transcriptase sequence (1–330 aa) of all the unique clones analyzed during the period 1998–2009. The scale corresponds to substitutions per base. Color images available online at
Surprisingly, the patient had undetectable VL under treatment from 1998 to 2002 despite intermediate resistance to AZT, and the potential high level resistance selection by single substitution in T215D, low-level resistance to 3TC, and the combination with a low genetic barrier drug such as EFV. Two additional RAMs were detected as single clones after first treatment: V106A was detected in the first sample from 2002, 1 month after treatment interruption, and Y188C was detected in 2008. Despite the confirmed combination in the same genome of the TAM-1 pattern and V106A or Y188C, the patient never experienced virological rebound while treated with AZT + 3TC + EFV, although it is highly likely that a longer period could have resulted in therapeutic failure.
The low genetic divergence of the sequences studied over more than 10 years of continuous monitoring is likely associated to the persistence of an R5 viral population with sustained RAMs and could be related to virological or host factors yet to be determined.
The results of this case are in agreement with recent evidence showing that most cases of infection through heterosexual transmission are monoclonal or oligoclonal and explain the prolonged persistence of RAMs in naive patients with TDR. Although this patient did not experience therapeutic failure, her initial treatment was clearly suboptimal as demonstrated by the detection of minor resistant mutants compromising NNRTIs in plasma samples posttreatment. Fortunately, this patient remains under sustained virological control with appropriate treatment despite the profile of drug resistance mutations. Nevertheless, this case stresses the importance of early detection and genotypic testing in all patients recently diagnosed with HIV-1 infection and the utility of more sensitive tools to detect relevant mutations occasionally present at low frequency.
Footnotes
Acknowledgments
J.L-G. is supported by a grant from the Instituto de Salud Carlos III, FIS Rio Hortega Program. The study has been supported by Grants FIPSE 36749, FIS-PI080806, and FP7 CARMUSYS PITN-GA-2008213592 to R.D.
Author Disclosure Statement
No competing financial interests exist.