
Abstract
HIV-1 epidemics in South America are believed to have originated in part from the subtype B epidemic initiated in the Caribbean/North America region. However, circulation of BF recombinants in similar proportions was extensively reported. Information currently shows that many BF recombinants share a recombination structure similar to that found in the CRF12_BF. In the present study, analyzing a set of 405 HIV sequences, we identified the most likely origin of the BF epidemic in an early event of recombination. We found that the subtype B epidemics in South America analyzed in the present study were initiated by a founder event that occurred in the early 1970s, a few years after the introduction of these strains in the Americas. Regarding the F/BF recombinant epidemics, by analyzing a subtype F genomic segment within the viral gene gag present in the majority of the BF recombinants, we found evidence of a geographic divergence very soon after the introduction of subtype F strains in South America. Moreover, through analysis of a subtype B segment present in all the CRF12_BF-like recombination structure, we estimated the circulation of the subtype B strain that gave rise to that recombinant structure around the same time period estimated for the introduction of subtype F strains. The HIV epidemics in South America were initiated in part through a founder event driven by subtype B strains coming from the previously established epidemic in the north of the continent. A second introduction driven by subtype F strains is likely to have encountered the incipient subtype B epidemic that soon after their arrival recombined with them, originating the BF epidemic in the region. These results may explain why in South America the majority of F sequences are found as BF recombinants.
Introduction
T
After being introduced into the American continent, the subtype B strains subsequently dispersed to the rest of America and to the rest of the world, initiating the different subtype B regional epidemics. However, several studies on molecular characterization have shown that HIV-1 epidemics in South America are constituted not only by subtype B strains but also by subtype F strains, as well as recombinants between both subtype B and F. 7 –18 In particular, the prevalence of BF recombinants is significantly higher than that observed for pure subtype F strains, reaching a prevalence similar to subtype B strains in some regions. 14 Considering the genetic distance between subtypes B and F, the presence of the BF recombinants in South America can only be explained by a second introduction of HIV-1 into the American Continent, independent of what occurred in the Caribbean/U.S. region with subtype B strains and, in particular, driven by subtype F strains. Current evidence about the origin of the BF epidemic was obtained from characterization of the recombinant structures. Those studies found that the majority of the BF recombinant sequences share common breakpoints that are identical to those found in circulating recombinant form 12 (CRF12_BF), 12 –16,19 which was the first recombinant to be identified in the region, suggesting a common BF recombinant ancestor. Later, new HIV circulating recombinant forms (CRFs) between subtype B and F, such as CRF28_BF and CRF29_BF, were identified in the region. 20 In addition, analysis of recombination patterns of BF recombinants circulating in South America has shown that ongoing current recombination drives a dynamic epidemic leading to the rise of new unique recombinant forms. 21
In the present study we analyzed the origin of the BF HIV epidemic and, in particular, that of the CRF12_BF that previous studies has proposed to be one of the most ancestral recombinant structures in the region. To assess this issue, we analyzed through phylogenetic methods a set of sequences from BF recombinants and subtype B strains that together span a significant proportion of the time period since the regional epidemics were established.
Materials and Methods
Sequences analyzed
Sequences analyzed in the present study were either obtained from the Los Alamos National Laboratory HIV sequence database (
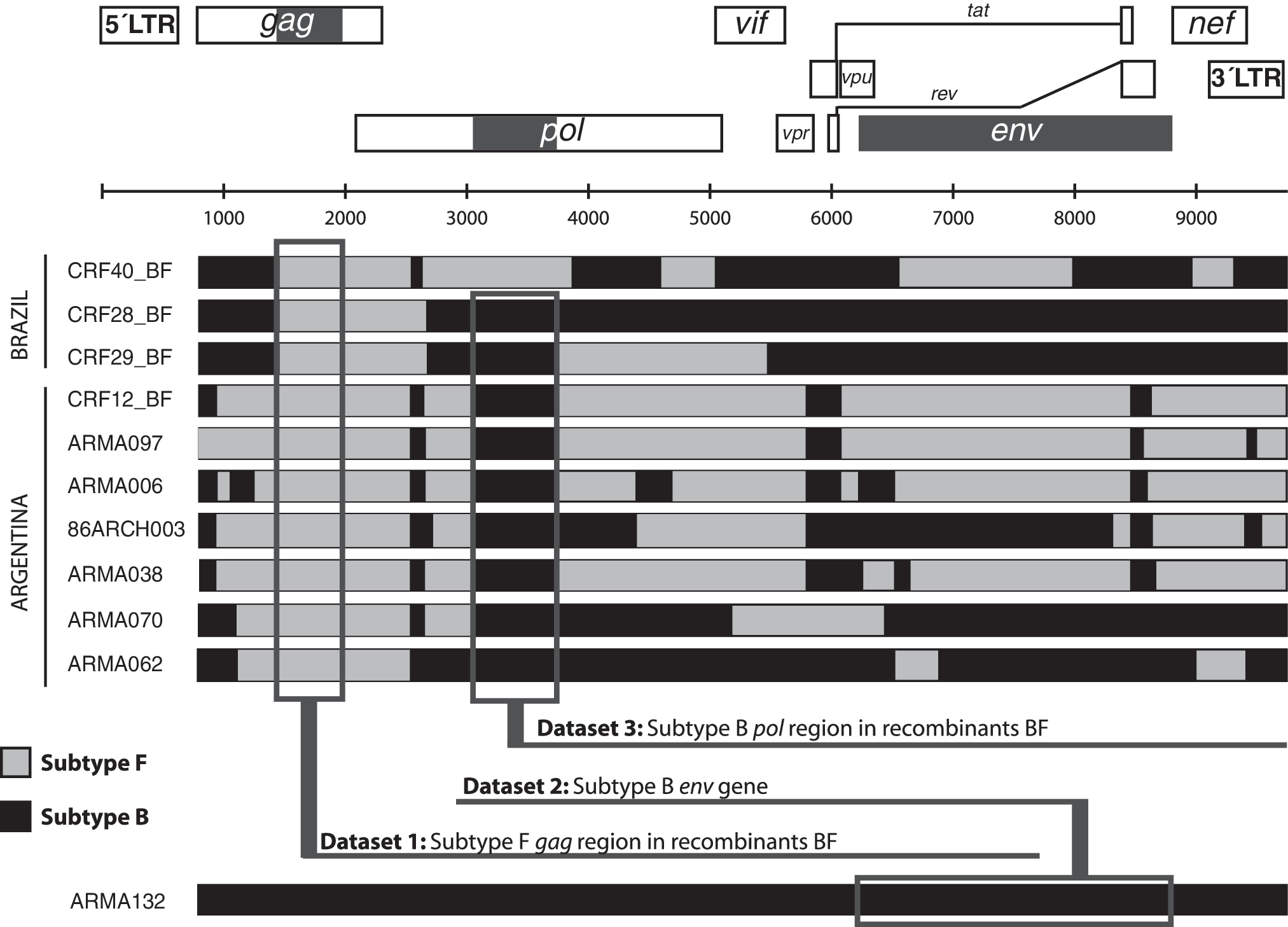
Viral genome regions under analysis. The recombination patterns of 10 full-length genomes previously characterized are shown as an example of diversity. The first three structures are recombinant forms circulating in Brazil. The fourth is the circulating recombinant form number 12. The other six structures are other unique recombinant forms identified in Argentina. One complete subtype B genome is represented by the sequence ARMA132. The three viral genome regions under analysis are highlighted in the schematic representation of the HIV genome (solid gray square) and in the sequences (open gray square). The gag region was analyzed in all the BF recombinants, the env region was analyzed in all the subtype B sequences, and the pol region was analyzed only in those BF recombinants that were subtype B in the region under analysis (in the example the CRF40_BF is excluded from this analysis).
For each dataset, alignments were performed by using Clustal W (BioEdit 7.0.4.1 sequence alignment editor
23
). Codon optimization was then performed with Gene Cutter [B. Gaschen (Los Alamos National Laboratory);
Viral subtype characterization
Sequences were aligned with subtype references sequences available (
The parsimony analysis of datasets
Phylogenetic trees based on parsimony method were obtained using TNT. 25 This software implements new technologies for dealing with large datasets 26,27 aimed to provide a thorough exploration of the tree space. This ensures that all the possible phylogenetic hypotheses that could be supported by the data are found. 27 Thus, any phylogenetic uncertainty present in the data is summarized in the corresponding strict consensus tree. Tree searches were performed as described elsewhere. 22 Briefly, we obtained 100 Random Addition Sequence trees, or Wagner (W) trees, 28 and the shorter ones were submitted to branch swapping using three rounds of a combination of Tree Fusing, Ratchet, Tree Drift, and Sectorial Search (W + T-R-Df-SS). 26 The best trees found in each W + T-R-Df-SS round were saved for ‘‘feeding’’ the subsequent W + T-R-Df-SS rounds (builder). Ten independent runs of the builder were performed and the best trees obtained from each builder were pooled and subjected to further cycles of T-R-Df-SS to verify that no further improvements of length were possible, at least under this experimental setting. Ambiguously supported branches were automatically collapsed during searches.
Estimation of the time to the most recent common ancestor
The BEAUti/BEAST v1.4.8 package [A. J. Drummond (University of Auckland, Auckland, New Zealand) and A. Rambaut (University of Edinburgh);
Statistical analysis
Chi-square test and Fisher's exact test were used to test the hypothesis of association between sampling country and phylogenetic clade.
Results
The phylogeography of subtype F
We first analyzed a total of 165 sequences of the gag gene from strains circulating in South America, previously characterized as BF recombinants, 22 in order to identify the different recombination patterns. We selected this dataset for analysis because it covers the broader range of years in the BF epidemic. After alignment and optimization the genomic viral region under analysis was 1147 bases in length. Results obtained through recombination analysis performed by bootscanning showed that the BF recombinant HIV sequences had one of either two different recombinant structures. Both structures had only one recombination breakpoint, although located in a different position: one of them had the recombination breakpoint around nucleotide 200 (989 in HXB2) in our alignment and the other one had the recombination breakpoint around nucleotide 500 (1301 in HXB2). The recombination pattern was found to be significantly associated with the country of sampling (p < 0.001, Fisher's exact test). All the sequences with a BF200-like recombinant structure came from Argentina, while 14 out of the 26 (53.8%) BF500-like structures came from Brazil. Comparative analysis of recombination breakpoints showed that the BF200 pattern is present in the recombination structure of circulating recombinant form 12 (CRF12_BF).
Next, in order to track the origin of the subtype F strain that gave rise to these BF recombinant forms, we performed a phylogenetic analysis over the larger region in which all the sequences belonged to the viral subtype F in order to avoid confounding effects due to recombination. The region analyzed was located between nucleotide positions 1401 and 1951 in HXB2 and constitute Dataset 1 (Fig. 1). After trimming the alignment, all the sequences were reanalyzed by bootscanning in order to confirm the absence of recombination breakpoints within the region, and the phylogenetic history was inferred by maximum parsimony and Bayesian methods. By using the first method, a total of 38 optimal trees was found and the strict consensus was constructed. As shown in Fig. 2A, this analysis indicated that there is a monophyletic clade that groups together all the sequences obtained in the American continent, whereas all the sequences collected outside the continent are paraphyletic. Within the American clade, two reciprocally monophyletic clades cluster with significantly different proportions (p < 0.001): the sequences from Brazil and the sequences from Argentina. Within the “Brazilian clade” only four out of the 25 (16%) sequences had a BF200-like structure, which, in addition, clustered in a monophyletic clade. In contrast, within the “Argentinean clade” the BF200-like structure was in the 48.5% (n = 67) of the sequences, the BF500-like structure in 12.3% (n = 17, five of them in a monophyletic clade), and the remaining 39.2% (n = 54) were pure subtype F sequences in the gag gene. Although the Bayesian method could not resolve phylogenetic relationships inside subtype F (Supplementary Fig. S1; Supplementary Data are available online at
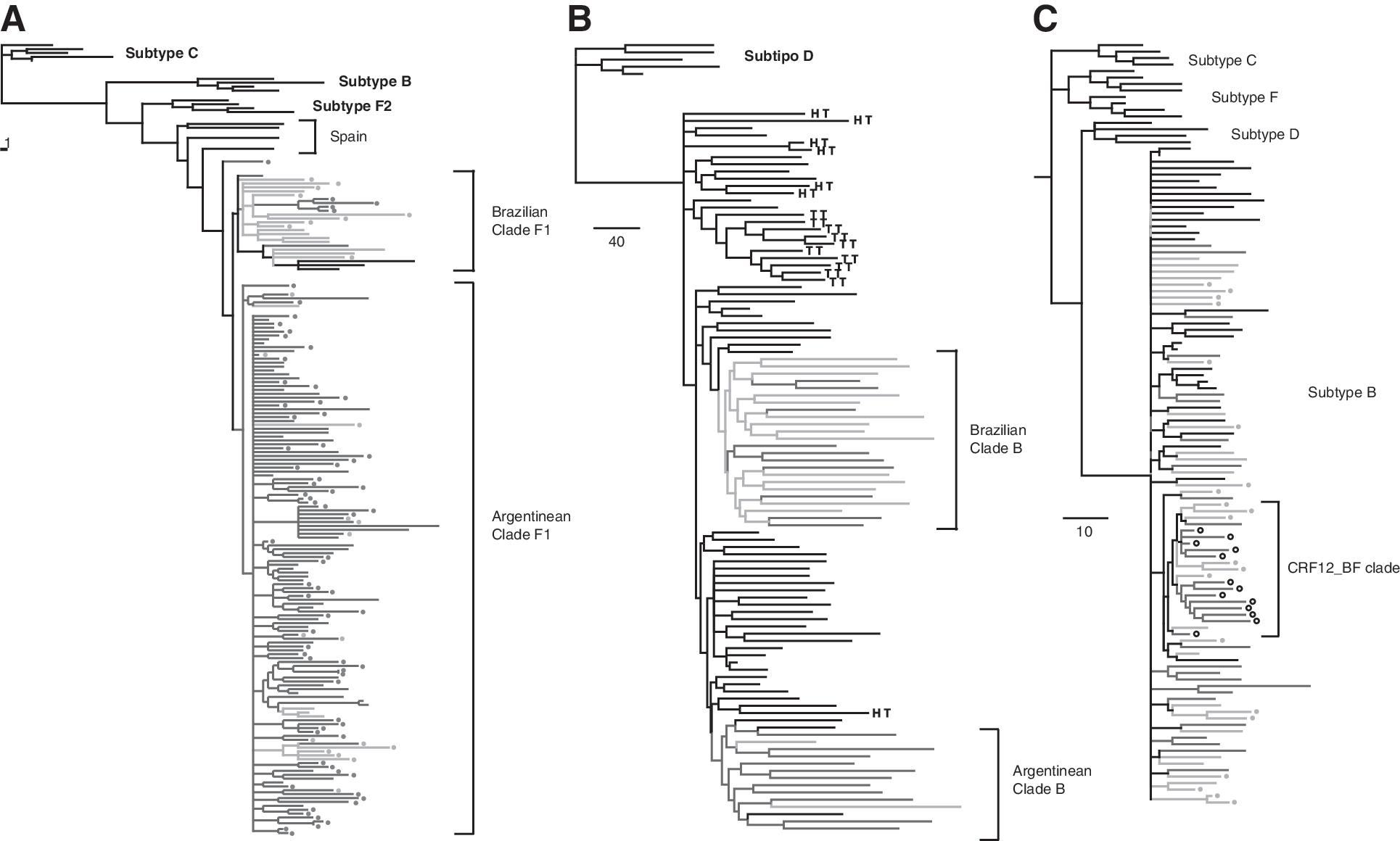
Phylogenetic reconstructions of regional HIV-1 epidemics. The analysis was performed by a maximum parsimony method. Branches are colored in green for samples of Brazilian origin and in blue for samples of Argentinean origin.
The origin of the subtype B epidemic in South America
To assess the origin of the subtype B epidemic in the region we performed a phylogenetic analysis over the env gene (Dataset 2, positions 6221–8795 in HXB2, Fig. 1). We based our analysis on the dataset previously described by Gilbert et al. 6 because it is the most complete evidence available for reconstruction of the subtype B epidemic in the American continent and included additional sequences previously obtained by our group and others obtained from Los Alamos Sequence Database. By using the parsimony method for phylogenetic reconstruction we were able to obtain four optimal trees. Different search strategies were not able to obtain shorter trees. As shown in Fig. 2B, we found that the majority of Argentinean and Brazilian sequences clustered in two reciprocally monophyletic clades. As for the subtype F sequences in dataset 1, character optimization suggests a Brazilian origin for one of them and an Argentinean origin for the other. The “Brazilian clade” has 14 sequences from Brazil and 10 sequences from Argentina. The “Argentinean clade” has 11 sequences from Argentina, 2 sequences from Brazil, and 3 sequences from the United States. The Bayesian-based phylogenetic reconstruction found the same two clades with a posterior probability of 1.00 and 0.78, respectively (Supplementary Fig. S2).
Phylogenetic reconstruction of subtype B regions in CRF12_BF resembles the monophyletic origin of subtype F
Considering previous reports that suggest that CRF12_BF is an ancestral recombination pattern we analyzed the larger subtype B segment present in the CRF12_BF and also in the majority of BF recombinants over a total of 102 sequences in order to assess the origin of the subtype B strain that originated the BF recombinant forms in the region. After gap striping, the region analyzed was of 500 bases in length, located within the pol gene between positions 3087 and 3597 in HXB2 (Fig. 1, dataset 3). After performing phylogenetic reconstruction through the parsimony method we obtained 49 optimal trees. As shown in Fig. 2C, the strict consensus has all the 13 CRF12_BF sequences clustering in a clade with other BF recombinants and subtype B sequences suggesting a monophyletic origin of subtype B genomic segments of CRF12_BF strains (“CRF12_BF clade”). The phylogenetic relationship for the remaining sequences could not be resolved and are collapsed in the node of subtype B. As shown in Fig. 3, the Bayesian-based phylogenetic reconstruction found the same clade in the majority-rule consensus tree with a posterior probability of 0.56 (see also Supplementary Fig. S3 and Supplementary Table S1).

Comparison of the phylogenetic reconstructions for the CRF12_BF clade obtained by the maximum parsimony and Bayesian method. CRF12_BF-like recombination structures are detailed with black branches and other BF recombinant structures are detailed in white. Subtype B sequences are detailed in gray. The viral subtype of the complete sequence is detailed in the first two letters of the name. The country of origin is detailed next with a two-letters code (AR: Argentina; BR: Brazil; US: United States).
Timing the origin of HIV epidemics in the region
Finally, considering the phylogenies reconstructed above, we aimed to estimate the time to the most recent common ancestor (TMRCA) for each of the subtype B and subtype F epidemics. Because the dating analysis is critically dependent on the monophyly of the clades for which the TMRCA is to be estimated, we performed the analysis only in those cases where that condition was satisfied according to phylogenetic support. The estimations of the TMRCAs and substitution rates were performed with the Bayesian Markov chain Monte Carlo (MCMC) phylogenetic analysis that allows the substitution rate to vary among branches in the tree.
In the case of the subtype F sequences we estimated the TMRCAs for the American subtype F clade, that is, for the ancestral node containing both “Brazilian clade” and “Argentinean clade” in Fig. 2A. Our analysis performed over dataset 1 found the maximum probability for the ancestor to that clade in the late 1960s/beginning of the 1970s (TMRCA: 1969; 95%HPD: 1959–1978, Fig. 4A). The majority-rule consensus tree obtained from the posterior sample of this analysis confirmed the monophyletic nature of this clade (Supplementary Fig. S1). In addition, we estimated the maximum probability for the time of divergence between viral subtypes F1 and F2 to be around in the late 1950s/early 1960s (TMRCA: 1956, 95%HPD: 1935–1972).
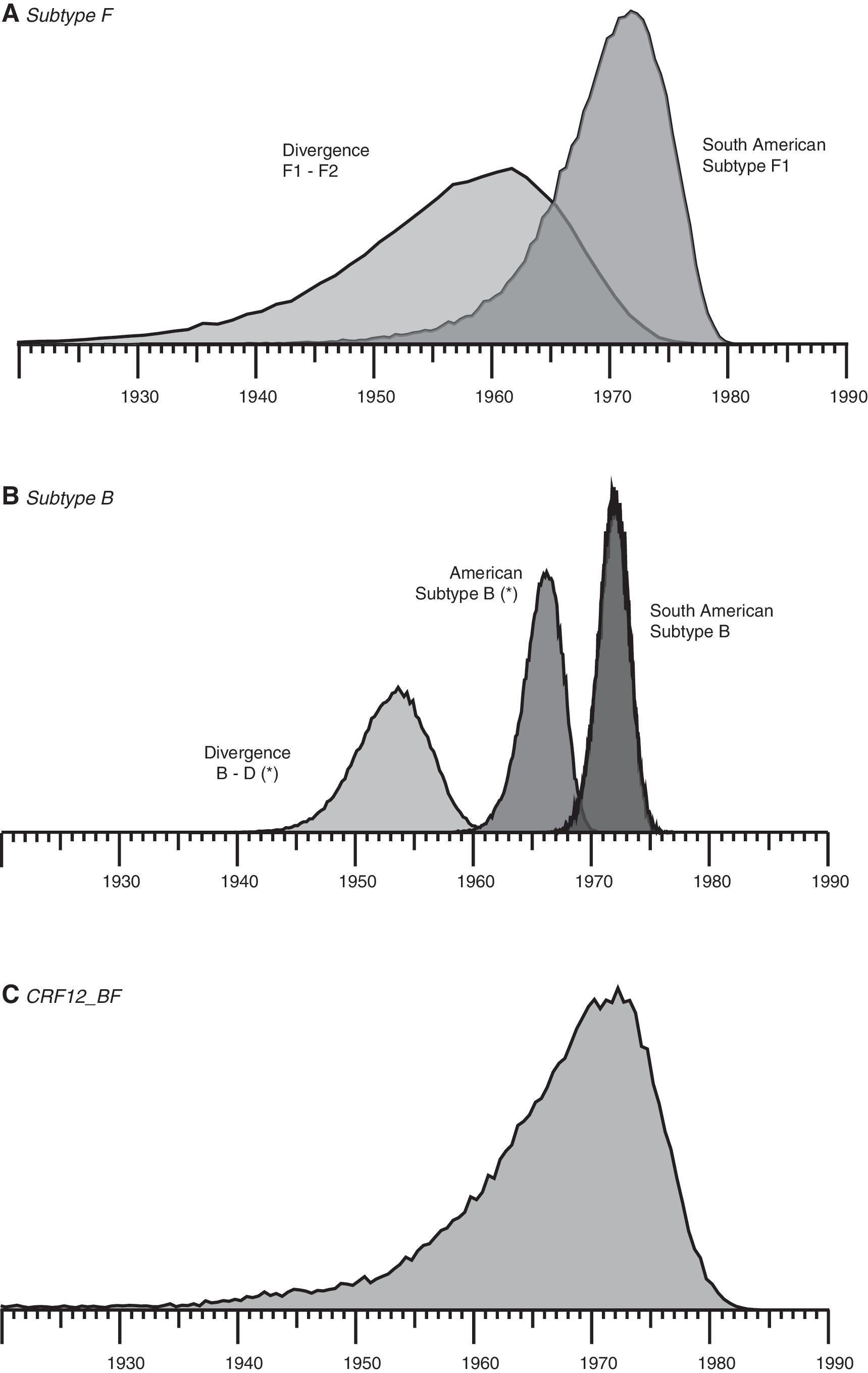
Estimation of the time to the most recent common ancestor (TMRCA). The density plots for the different TMRCAs estimations are shown. (
In the case of the subtype B sequences we estimated over dataset 2 the TMRCAs for both the Argentinean and Brazilian clades shown in Fig. 2B. Our analysis found identical results for both clades: located at 1972 (95%HPD: 1970–1974) for the Argentinean clade and 1972 (95%HPD: 1969–1974) for the Brazilian clade (Fig. 4B). The majority-rule consensus tree obtained from the posterior sample of this analysis confirms the monophyletic nature of these two clades with a posterior probability of 0.78 and 1.00, respectively (Supplementary Fig. S2). Regarding the substitution rates, we found a rate of 0.00419 (0.00374–0.00464) substitutions/site/year for the env gene of subtype B and 0.00167 (0.00115–0.00210) substitutions/site/year for the gag gene of subtype F.
Finally, we aimed to estimate the TMRCA for the CRF12_BF by analyzing dataset 3. In this case, the maximum parsimony tree differed, within the “CRF12_BF clade,” from the Bayesian tree in one sequence: as shown in Fig. 3 the recombinant BF sequence BREPM12609 (Accession number: DQ085873) was located outside the “CRF12_BF clade” in the maximum parsimony tree. Also, we found within this clade, a more resolved phylogeny when the parsimony method was applied compared to that obtained with the Bayesian method in which the majority of nodes were collapsed. In spite of this, the Bayesian method still resolves the “CRF12_BF clade” and therefore allows us to estimate the TMRCA. By carrying out this analysis we estimated that the maximum probability for the most recent common ancestor of the CRF12_BF was the early 1970s (TMRCA: 1969, 95%HPD: 1946–1981, Fig. 4C).
Discussion
In the present study we looked for an explanation of the particular HIV-1 epidemic that is present in South America where, for many years, several studies have consistently reported the cocirculation of subtype B strains and BF recombinants but a very low prevalence of pure subtype F strains. 10,12 –16,30,31 Because we were analyzing the origin of the epidemic in South America and considering that the first detected cases of AIDS in the region occurred in Brazil and Argentina, we looked mainly for sequences derived from those two countries. In addition, the number of HIV sequences from other countries is very limited. The similarity in the recombination structures of the BF sequences suggested a common recombinant ancestor, but that hypothesis was never exhaustively evaluated before. The advantage of having, on the one hand, sequences of both subtype B and recombinants BF recovered from stored plasma of the past 20 years in our region and, on the other hand, an accurate phylogenetic reconstruction of subtype B in the Americas obtained by Gilbert et al. 6 allowed us to dissect the most ancestral phylogenetic relationships in the initiation of the recombinant BF epidemic.
Because TMRCA calculations critically depend on the monophyletic nature of regional epidemics and considering that monophyletic clades could be a consequence of the lack of sequences that, if included, could split the identified clusters, we included in this study the most representative set of sequences available. In this sense, the subtype F and recombinants BF sequences analyzed were obtained from plasma samples stored from 1987 through 2006 in five different hospitals in Argentina that function as voluntary and counseling testing (VCT) sites.
Because VCTs are considered one of the preferred sources of samples for epidemiological studies over the general population, 32 we expect those sequences to be a representative sample of the local epidemic. On the other hand, sequences obtained from two cross-sectional prevalence studies performed over two at-risk cohorts of MSM 33 and female sex workers 31 designed to obtain a representative sample of each group were also included. Sequences from other regions were limited to the availability in databases. However, in all the monophyletic clades analyzed, we found some level of heterogeneity in terms of the sequence origin, suggesting that we are likely analyzing unbiased samples of viral sequences. The same applies to subtype B sequences, with the exception that for this case we also added those sequences previously described by Gilbert et al. 6 because previous epidemiological data suggested that the local epidemics in the American continent were related. In fact, it is known that one arm of the global dispersion of HIV began by the introduction in the Caribbean/U.S. region of subtype B strains that subsequently dispersed to the rest of America and to the rest of the world. 5,6,34 In the particular case of CRF12_BF, the 13 complete genomes sequences were obtained from three different sources: five of them were obtained from the cross-sectional study of prevalence in female sex workers mentioned above, 31 three of them from a study of pregnant women seeking HIV testing, 15,35 and five of them from a study of drug resistance over primary and chronically infected patients who attended to a VCT site. 12,36 Therefore, although the number of complete CRF12_BF genomes available is limited, in the phylogenetic reconstruction it is likely that they represent distantly related sequences within a hypothetical group of all the CRF12_BF circulating.
The fact that all the South American subtype B sequences grouped in only two monophyletic clades with a geographic correlate, which is also the case for subtype F sequences, suggests that these epidemics are predominantly fed by local events of transmission even when they occur in populations of geographically close regions as is the case of Argentina and Brazil. The presence of sequences from Brazil in the Argentinean clade and sequences of Argentina in the Brazilian clade shows that circulation among regions actually occurs as recently suggested, 21 although the monophyletic nature of both clades suggests an independent origin by one or a few related strains.
Previous studies have reported the initiation of HIV epidemics through a founder effect in San Francisco 37 and Trinidad and Tobago, 38 and other regions of the world, 39 –43 and in our study this may be explained by a divergence of the subtype F strains in both populations at the very early moment of the colonization, after their introduction in South America probably from Africa, as subtype F1 is not found in other regions of the world. For subtype B, considering our results, a likely explanation would be that one or a few related strains imported from the subtype B epidemic established in North America successfully spread and established these local epidemics. Taking into account that previous reports have shown that HIV transmission is primarily attributable to HIV-vulnerable groups, 44 i.e., concentrated epidemic, 45 we consider a possible explanation for the fact that a few strains successfully spreading over others that finally were limited. Also, the dates estimated for the TMRCA of these local epidemics in South America suggest that there had been a significant number of autochthonous infections by the time the first cases started to be identified.
Our estimations for the Brazilian clade subtype B are consistent with those previously reported by Bello et al. 46 and almost identical to our estimations for the Argentinean clade B, suggesting that the introduction of subtype B in Argentina and in Brazil occurred almost at the same time and very soon after the introduction of subtype B in America. Based on these results, it is likely that HIV strains that circulate in different regions may have differentiated genetic characteristics even when they belong to the same viral subtypes and the differences may be attributable to founder events in the origin of the epidemics in different regions. In this sense, it would be interesting to perform phylogeography studies of HIV epidemics in other regions of the world to evaluate whether the majority of them were initiated by founder events and to better understand the genetic characteristics of strains currently circulating.
In addition, the fact that the subtype B regions of the different CRF12_BF-like recombinant genomes have a common ancestor closer to themselves than to any other subtype B sequences suggests that an event of recombination occurred in the very early years of the epidemic generating a recombinant form, subsequently identified as CRF12_BF. In fact, when we analyzed the subtype B region of the BF recombinants, we found them to have a TMRCA distribution similar to that estimated for the American subtype F strains (Fig. 4). This means that the subtype B strain that originated the CRF12_BF by recombination with a subtype F strain was circulating in the region at the same time as the subtype F strains arrived to America. Therefore, it is likely that the recombination events occurred in the early moments of the initiation of the F/BF epidemic. This is in accordance with the observation that subtype F is almost absent in the region while the majority of non-B strains are recombinant BF and also with the fact that the majority of BF recombinants share similar patterns of recombination highly related to those found in CRF12_BF. 12 –16,19
Regarding the estimated evolutionary rates, our estimation of around 0.00162 substitution per year per site for the gag dataset is similar to that obtained for the gag gene by Korber et al. 5 (0.0009–0.0027) and our estimation of around 0.00419 for the env gene is also similar to that estimated for env by Leitner et al. 47 (0.0046–0.0088) and Robbins et al. 34 (0.00473), but higher than that obtained by Korber et al. 5 for env (0.0018–0.0028). The last difference is likely to be due to the fact that posterior studies demonstrated that a relaxed molecular clock better explains the phylogenetic reconstruction of HIV sequences. In this sense, we also found a significant variation in evolutionary rates among branches in the tree as shown by the standard deviation of the uncorrelated lognormal relaxed clock found in our analysis (ucld.std 95%HD: 0.195 ± 0.037 for env gene B and 0.456 ± 0.076 for gag gene F). Recently, Abecasis et al. 48 estimated the evolutionary rates for the different HIV-1 subtypes, obtaining a rate for subtype B very similar to ours when analyzing the env gene [a mean rate of 0.0045 substitutions/site/year compared to the 0.00419 (0.00374–0.00464) estimated by us]. In addition, while Abecasis et al. 48 found subtype G to have the higher evolutionary rate compared to subtypes B, C, A1, D, and recombinants AG and AE, contextualizing our estimations in these set of results, subtype F in the gag gene seems to have an evolutionary rate similar to that observed for the pol gene in the other HIV-1 subtypes [0.00167 (0.00115–0.00210) compared to a rate between 0.001 and 0.002 for the majority of the other subtypes].
Finally, it is important to remember that the HIV-1 epidemic in South America is also composed by a minority, although still an important proportion, of subtype C infections. In the present study we limited our analysis to both subtype B and subtype F/recombinant BF epidemics because those are the viral subtypes found in the oldest samples obtained from patients in the region. In fact, a later initiation of the subtype C epidemic in South America compared to the subtype B epidemic was previously reported 49 with an estimated initiation at the beginning of the 1990s. The higher prevalence of subtype B and recombinants BF compared to subtype C infections may be a consequence of an earlier initiation of the first ones, although additional factors influencing the evolutionary rate of circulating strains, such as the significantly higher rate estimated previously for subtype C strains in Brazil, 49 make the current scenario not necessarily stable and may lead to a shift in the future of the epidemic.
Conclusions
In conclusion, by applying parsimony and Bayesian methods to the reconstruction of the evolutionary history of HIV in America we were able to dissect the origin of regional HIV epidemics after their introduction in the Americas. According to our results subtype B strains of HIV coming from the north of the Americas arrived in South America in the early 1970s and established the South American subtype-B epidemic through a founder event.
The relative short period of time between the estimated origin of the U.S. subtype B epidemic and the South American U.S.-related subtype B epidemic suggests rapid spreading of the infection to the whole American continent after successful colonization through the Caribbean/U.S. region. At the same time a second wave of HIV strains arrived from Africa and established the subtype F epidemic of South America. Soon after the introduction of subtype F strains in this region, it is likely that a recombination with a subtype B strain had occurred giving rise to a recombinant form with a CRF12_BF-like recombination pattern. This founder recombinant event established a BF epidemic, giving rise to other unique recombinant forms by further recombination with subtype B strains. Finally, our results suggest that by the time the first cases of AIDS were identified, not only the Caribbean/North American HIV epidemic but also several regional HIV epidemics in South America were already established.
Footnotes
Acknowledgments
We thank Dr. Michael Worobey, Department of Ecology and Evolutionary Biology University of Arizona, for kindly providing the dataset for env analysis previously published by his group. DAD acknowledges the support from the Fogarty International Center/NIH grant through the AIDS International Training and Research Program at Mount Sinai School of Medicine-Argentina Program: Grant D43 TW001037.
Author Disclosure Statement
No institutional of commercial affiliations that might pose a conflict of interest regarding the publication of this manuscript exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.