
Abstract
We genetically characterized the extent of variation in HIV-1 LTR sequences from 11 mother-to-child transmission (MTCT) pairs from HIV-1-infected individuals from North India. Nine pairs were found to be infected with subtype C virus whereas two pairs were infected with subtype B virus. They harbored the characteristic three and two NF-κB sites, respectively. The analysis of intrasubtype divergence between B and C revealed greater diversity with subtype B LTR sequences than subtype C (p < 0.005). Significant evolutionary divergence of subtype C and subtype B was found in NFAT-III (p < 0.000001), NFAT-II (p < 0.0001), and USF (p < 0.005) transcription factor binding sites (TFBS). NF-κB-I, Sp I and II, Ets-I, AP-I and II, and TATA Box TFBS were highly conserved in both the subtypes. An alternate secondary structure of Tar was detected in the VT5 sample due to the point mutation from G to C (position +21) and T to C (position +38).
A
HIV-1 vertical transmission occurs mainly at three stages: prepartum (transplacental), intrapartum (exposure of infant's skin and mucus membrane to maternal blood and vaginal secretions), and postpartum (breastfeeding). 3 The various factors involved in successful transmission include low CD4 T cell count, high virus titer in maternal blood, advanced stage of disease progression, maternal immune response to HIV-1 antigens, along with the presence of other sexually transmitted diseases during pregnancy. 3
HIV-1 exploits host signaling pathways and viral regulatory proteins synergistically for its transcriptional regulation through its long terminal repeat (LTR) region, present at both terminals (5′-LTR and 3′-LTR) of its genome. The HIV-1 LTR quasispecies are approximately 640 bp in length and like other retroviral LTRs, are segmented into U3, R, and U5 regions. 4 The U3 region of the 3′-LTR, which overlaps with the Nef-coding region, has several sites that bind cellular transcription factors and is further subdivided into the modulatory, enhancer, and core/promoter functional regions. 5 The U3 region is of great interest because of the presence of transcription factor binding sites (TFBS) such as nuclear factor-kappa B (NF-κB), activator protein 1 (AP1), Sp, nuclear factor of activated T cells (NF-AT), T cell-specific transcription factor-1alpha (TCF-1α), and the TATA promoter, which regulate HIV transcription. Previous studies showed that Nef/LTR quasispecies were under greater selective pressure than Nef alone, and immune selection pressure acting on Tat and Nef, overlapping the region of 3′-LTR, may directly influence LTR diversity as 5′-LTR does not encode proteins capable of interacting with the immune system. 6 Among multiple cellular factors known to bind to the HIV LTR, Sp proteins appear to be crucial for basal-level HIV gene expression 7 whereas the NF-κB protein is induced in activated T lymphocytes and enhances HIV gene expression. 8
Intersubtype LTR recombinants involving subtype A, C, and D subtype-specific genomes were earlier reported among Tanzanian infants. 9 Rodrigues et al. carried out genetic and functional studies on the characterization of the HIV-1 5′-LTR regions of subtype A and C from samples collected from Pune (Maharashtra state) and Calcutta (West Bengal state) located in Central and Eastern India, respectively. 10 We recently reported the predominance of unique subtype B LTRs and two novel mosaic HIV-1 LTR B/C recombinant genomes from the Chandigarh region of Punjab, North India. 11
To the best of our knowledge, to date, no data on genetic variations of LTR quasispecies associated with MTCT in India exist. In this study, we characterized HIV-1 LTR quasispecies from 11 mother–child pairs following vertical transmission from different regions of North India (Punjab, Haryana, and Himachal Pradesh). These states are located just north of Delhi, the capital of India.
Blood samples were collected from a North Indian cohort of 11 mother–child pairs from different regions of North India attending the Immunodeficiency Clinic of Post Graduate Institute of Medical Research and Education, Chandigarh. The CD4 cell count was enumerated by a bead-based FACS count kit (BD Biosciences Immunocytometry System, San Jose, CA). Genomic DNA extraction, polymerase chain reaction (PCR) amplification, and sequencing were carried out as described by us previously. 11 All PCR reactions were performed with hi-fidelity Taq polymerase (Invitrogen). Mother and infant samples were processed separately to avoid potential cross-contamination. At least two to four clones were genetically analyzed for each sample and only the representative sequences were analyzed.
Analyses of HIV-1 subtypes were performed using the RIP 3.0 (
To explain the strong, environmentally driven fitness of point mutations in the HIV-1 LTR promoter, we examined the affinity of the DNA binding sites. For this we used a web-based program, Matinspector (
Cohort characteristics were as follows. The mean age of the mothers was 29 ± 4 years and of the children was 7 ± 4 years when the blood was collected (2007–2009). The median CD4 count for mothers was 249 (range 46–497) and for children it was 870 (136–1487). Among the children 82% were male and 72% were on antiretroviral treatment (ART). In addition, 54% of mothers were also on ART. Four patients were coinfected with CMV retinitis (VT6), herpes zoster (VTD2), and tuberculosis (VTE2 and VTE22). The mode of transmission among the mothers (except one, NII-PGI-IND-VTD4) was confirmed as heterosexual transmission after counseling. All the transmissions among children were confirmed as vertical transmission, as sex abuse was not reported by the parents during counseling.
Phylogenetic analysis of 11 mother–child consensus LTR sequences showed that 9 of 11 pairs formed a subcluster segregating from Indian subtype C clusters and pairs 5 and 7 in subtype B cluster (Fig. 1). The consensus sequence from five pairs (pairs 1, 6, 7, 9, and 11) formed a monophyletic clade indicating an epidemiological linkage of these samples. Pairs 3, 4, and 8 grouped together in a confined subtree, indicating that the variants from the mother–infant pairs are closer to each other. The consensus LTR for pairs 2, 5, and 10 formed a nonsignificant grouping. This kind of result was previously observed in a Tanzanian mother–child cohort. 6 We also observed in a single pair (pair 5, VTD4 and E4) that the child LTR quasispecies (sample taken at 11 years) was more diverse than that of the mother, suggesting transmission of multiple maternal variants or multiple infant infections with HIV-1. The transmission of multiple variants may be a due to the prolonged exposure in utero, the maternal viral load during delivery to which the child was exposed, or subsequent reinfection of the infant during breastfeeding. 6 The extent of the diversity correlated with the age of the children. It was previously proposed that the minor HIV-1 genotype predominates initially as a homogeneous population in the infant, which then becomes diverse as the infant grows older. 3

Phylogenetic analysis of North Indian isolates following MTCT. A neighbor-joining tree was constructed in MEGA4 with reference sequences downloaded from the HIV Los Alamos database. The accession numbers of the reference sequences are given in parentheses. Filled numbers belong to children whereas open numbers belong to mothers.
The analysis of intrasubtype diversity at comparable mother–child LTR sequences showed that the subtype B population is more diverse than the subtype C population (p < 0.005; Fig. 2A). The nucleotide distances among the mother–child samples were greater in pair 5 followed by pair 11 (Fig. 2B). We then calculated the maximum composite likelihood estimate of the pattern of nucleotide substitution between the mother–child of pair 5 (D4 and E4) consensus LTR sequences, and found that rates of different transitional substitutions compared more to transversional substitutions (p < 0.005, Table 1).
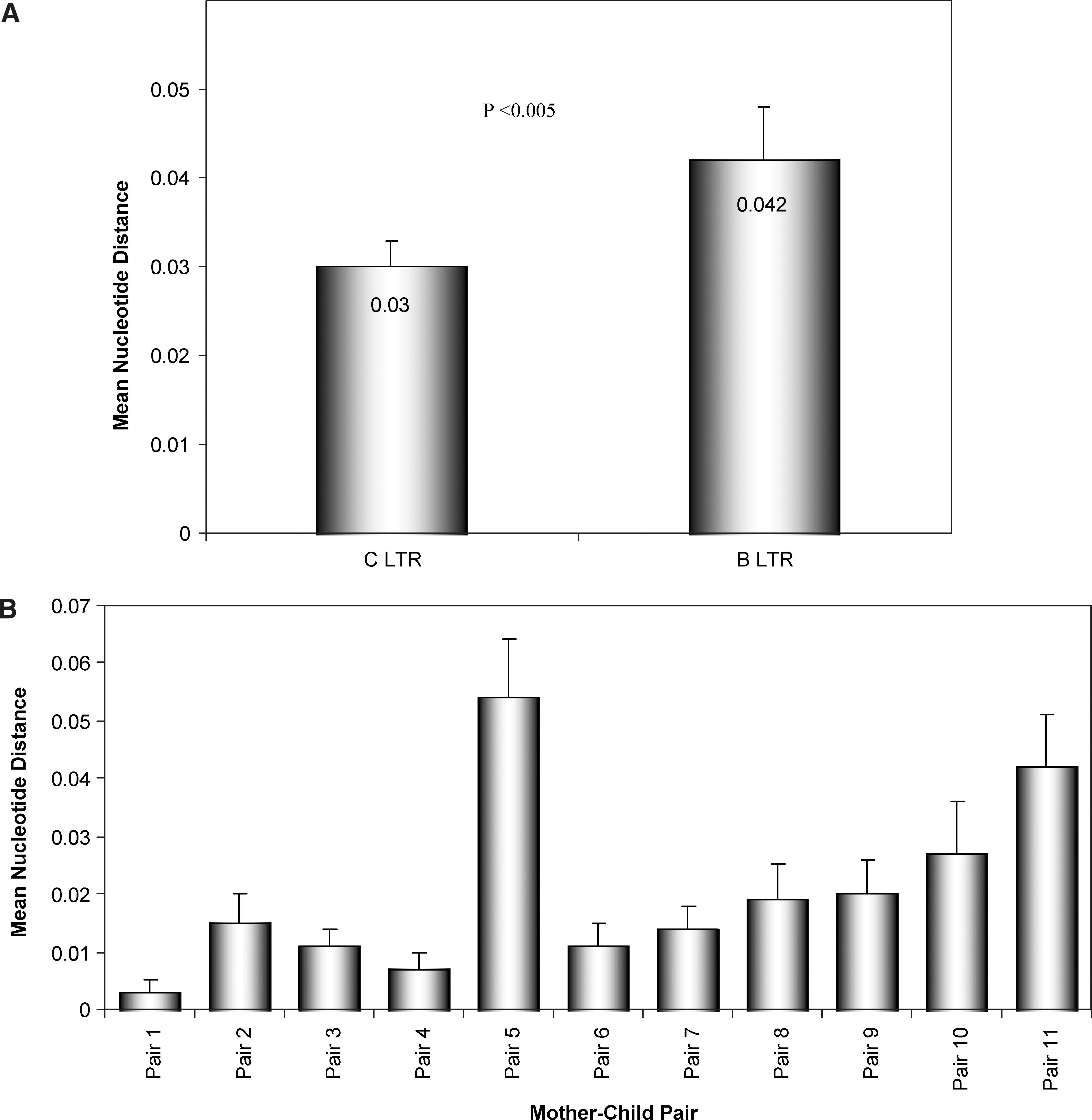
Mean nucleotide distance between subtype B and C LTRs from North India. Nucleotide distance estimations were performed using two substitution models (Kimura two-parameter and p-distance) in 100 bootstrapped data sets using MEGA4; only the p-distance model is shown. The analysis of diversity of subtype B LTR includes our previously published B LTR sequences. 11
Each entry shows the probability of substitution from one base (row) to another base (column) instantaneously. Only entries within a row should be compared. Rates of different transitional substitutions are shown in bold and those of transversional substitutions are shown in italics. The transition/transversion rate ratios are k 1 = 3.18 (purines) and k 2 = 1.482 (pyrimidines). The overall transition/transversion bias is R = 1.225, where R = [A · G · k 1 + T · C · k 2]/[(A + G) · (T + C)].
The TFBS analysis suggests that the HIV-1 LTR region is densely packed with overlapping protein binding sites, with 96–113 conserved potential sites in 86% (common TFBS found at least 19 of 22 sequences) of the sequences derived from different MCTC samples in North India. This suggests that the point mutations potentially change number of TFBS, which also provide the specificity of the host cellular transcription factors in vertical transmission. The characteristic polymorphisms of the NF-κB sites (two in subtype B and three in subtype C) were also detected in north Indian isolates of subtype B and C along with clade-specific and region-specific conservation in the promoter (TATAA), the enhancer (three Sp and two/three NF-κB sites), and the modulatory region (two AP-1, three NFAT and Ets-1, and one TCF-1α).
Despite their role in the interaction with host and viral regulatory factors, we observed a high genetic heterogeneity in the sequences involved in transcription regulation located at the U3 region in the Indian subtype B population. Subtype B exhibited higher nucleotide diversity when compared to subtype C at NFAT-III, II, I, and USF TFBS (Table 2). A significant evolutionary divergence of clade C and clade B was found in NFAT-III, 0.017 vs. 0.071 (p < 0.000001), NFAT-II, 0.015 vs. 0.067 (p < 0.0001), and USF, 0.038 vs. 0.091 (p < 0.005). NF-κB-III in subtype C, NF-κB-I, Sp I, and II are the highest conserved TFBS in both the subtypes along with Ets-I, AP-I and II, and TATA Box (data not shown).
Mutant frequency has been calculated by dividing the number of mutations with respect to the reference HXB2 (for Subtype B; n = 4) and Indian Consensus C (for Subtype C; n = 18) by the total number of nucleotides sequenced in each TFBS as described previously. 14 Evolutionary divergence has been calculated in MEGA 4.1. The number of base differences per site from averaging over all sequence pairs is shown.
A stem–loop structure called Tar (Fig. 3A. numbered as +1 to +59) is important for the Tat protein interaction. Three-nucleotide-long bulge (TCT) and 6-nucleotide-long (CTGGGA) loop regions were conserved in 21 of our samples. Twenty-one of 22 isolates retained the intact bulge and loop regions as well as the six principal base nucleotides involved in supporting the (GGG and CCC) bent rod TAR RNA secondary structure (nucleotides +1 to +3 and +57 to +59). A deletion of G was found in VTE8 in the 5′-base region. Some point mutations were observed in isolates VT3, VT4, VT5, VTD23, VTE23, VTD24, VTD25, and VTD26 compared to the consensus C and Indian prototype subtype C–93IN905 sequence. The sequences of subtype B Tar were mostly conserved when compared to the HXB2 sequence.
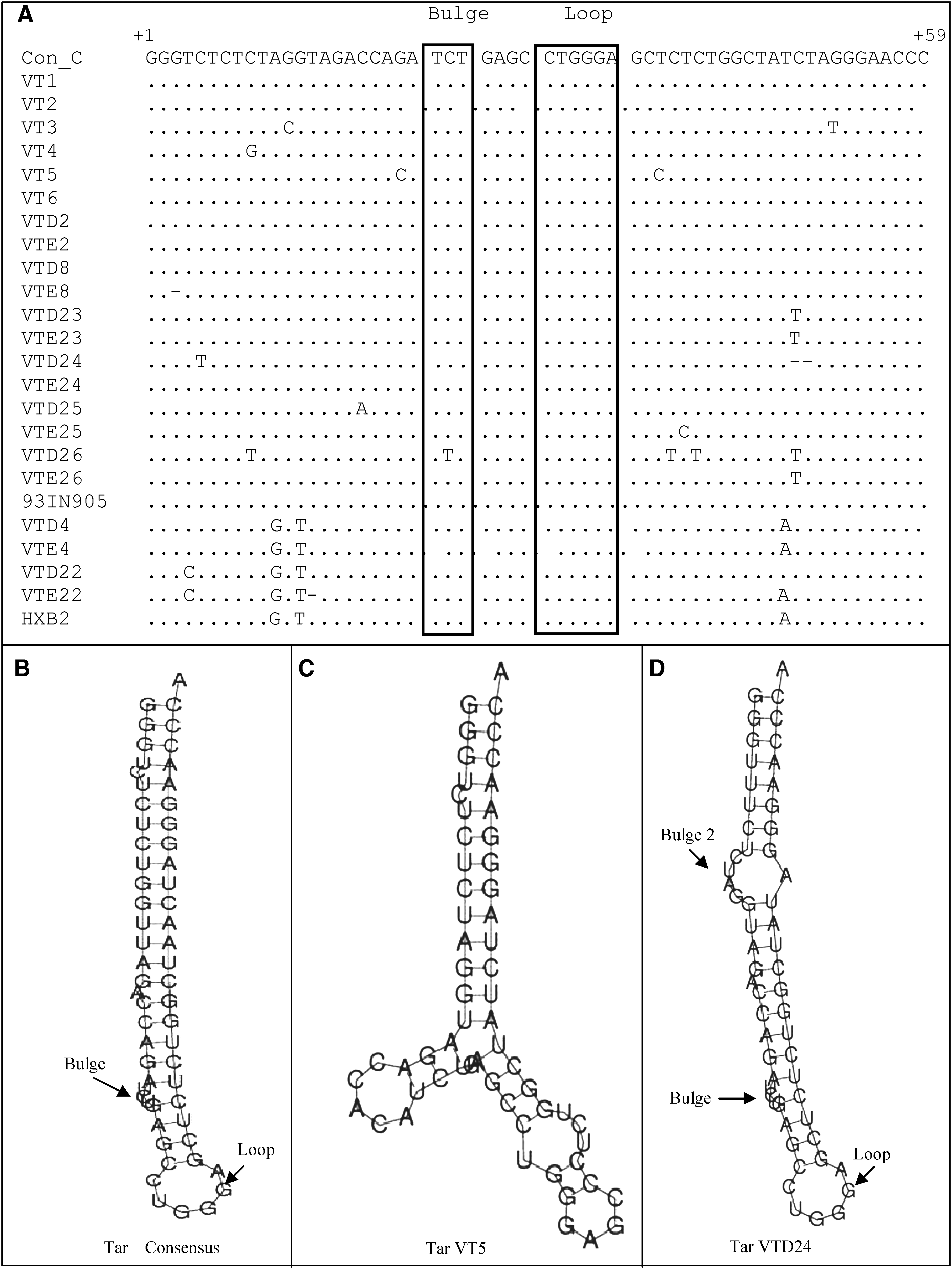
Sequence variations and secondary structure of TAR. (
We then carried out in silico RNA structure predictions using two different approaches, conventional single-sequence algorithms using mFold (
To our knowledge, this is the largest number of mother–child pairs studied to date in India, the third largest HIV-1 epidemic in the world, and the first study to analyze LTR variants associated with MTCT here. We describe the HIV-1 LTR subtype-specific variability in MTCT among viruses circulating in India. We also observed monophyletic and epidemiologically linked LTR sequences from North India. It was previously reported that the HIV-1 genetic architecture in India is related to African strains, but it has its own genetic makeup to create a monophyletic tree segregating from the HIV-1C clade.
We observed that the majority of subtype C (9 pairs) and subtype B (two pairs) successfully transmitted from mother to child. Previous comparative studies were from different non-subtype B HIV-1 LTR sequences including subtypes A, C, D, E, F, G, H, and J with subtype B showing a high degree of genetic diversity. High genetic diversities in NFAT-1, Sp, and USF TFBS were observed in non-subtype B compared to subtype B-specific TFBS in LTRs. But in north Indian strains subtype B is a more diverse TFBS than subtype C. The highest genetic diversity was observed in pair 5 and pair 11. The diversity in pair 5 correlated with the age of children (11 years), but not in pair 11 (age of the children 2 years and the youngest one in the study cohort). It was previously proposed that the minor HIV-1 genotype predominates initially as a homogeneous population in the infant and then becomes diverse as the infant grows older. 3 In view of the lack of a viral load assay, we choose the CD4 count as an indicator of transmission, which was used previously, 15 but its measurement is independent of viral subtypes and cannot be used as absolute predictors of MTCT of HIV-1. 16
In conclusion, we have for the first time genetically analyzed the diversity of LTR sequences in mother–child pairs from North India and studied its impact on the predicted secondary structure. The diversity in mother–child LTR variants showed some correlation with the age of the children; however, other undefined viral characteristics may also be responsible for the viral diversity in children. The monophyletic nature of the phylogenetic tree observed with LTR sequences showed correlations with previous reports of other HIV-1 gene segment analyses from India. The environmentally driven fitness and immune pressure exerted on the HIV-1 nonstructural region may also play an important role in vertical transmission. The analysis of intrasubtype divergence between B and C in North India showed that the subtype B LTR is more diverse than the subtype C LTR (p < 0.005), which has been observed for the first time and is the major finding, with enormous implications for gene expression and replication.
GenBank Accession Numbers
Consensus sequences generated from each mother–child pair were submitted to GenBank under the following accession numbers: FJ432056–FJ432067 (pairs 1–6) and GQ215489–GQ215498 (pairs 7–11).
Footnotes
Acknowledgments
This work was supported by grants from the Department of Biotechnology, Indian Council of Medical Research, Government of India. Genetic clones 93IN905 and pNL4-3 were obtained from the AIDS Reference and Reagent Program of the NIH (Bethesda, MD). The authors would like to thank Karthika Arumugam, St. John's National Academy of Health Sciences, Bangalore for her help with statistical analysis. Also, the help received from Dr. Surjit Singh, PGIMER, Chandigarh, in clinical characterization of mother-child samples is gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.