
Abstract
R5 and X4 HIV strains use CCR5 or CXCR4 chemokine receptors (CKRs), respectively, for entry. Preferential growth of X4 vs. R5 HIV in cell lines reflects constitutive expression of CXCR4, but not CCR5 (in contrast to dual expression on primary T cells), and CXCR4 is the predominant CKR found on most tumors. Non-Hodgkin's B cell lymphomas (NHL) are increased among HIV+ patients, and interactions between HIV envelope and CKRs may contribute to lymphomagenesis. Despite strong evidence for a CXCR4–SDF-1 oncogenic axis, no in vitro evaluation of CXCR4-mediated normal lymphocyte transformation has been published. Exposure of normal B cells to EBV in the presence of X4 gp120 (but not R5 gp120) increased proliferation and BLCL outgrowth, comparable to anti-CD40 mAb costimulation. This suggests a role for X4 tropic viral envelope signaling via CXCR4 and/or CXCR7 in HIV-associated lymphomagenesis.
R5
We 9 and others 10 showed that X4 gp120 can bind CXCR4 on PBMCs and transduce chemotactic signals, independent of CD4. Subsequently, gp120 engagement of CXCR4 on T cells was found to trigger Rho and cyclophilin-dependent cytoskeletal phosphorylation and nuclear translocation. 11 Another SDF-1 receptor, CXCR7, is necessary for sustained EBV+ B lymphoblastoid cell line (BLCL) proliferation, and is more strongly induced by EBNA2 from Type 1 vs. Type 2 EBV, apparently explaining the latter's poorer transforming efficiency. 12 Thus, gp120 signaling via CXCR4 or CXCR7 could be implicated in the increased B cell lymphomagenesis seen among HIV+ patients. 13
Cross-sectional studies in HIV+ adults 14 showed a correlation between the SDF-1-3'A variant and NHL. Intriguingly, prospective studies 15 correlated increased SDF-1 mRNA with subsequent NHL among HIV+, but not uninfected, children. Conversely, CXCR4, can mediate apoptotic signals, depending on the kinase pathways concomitantly activated, 16 and X4 HIV strains and envelope have been implicated in Fas-mediated and Fas independent autophagy-mediated CD4 cell death of uninfected cells. 16 –20 However, the latter pathway requires fusogenic gp41 rather than gp120 CKR signaling, and is efficiently mediated by both R5 and X4 virus after CD4 binding. 20 CCR5 may also mediate HIV-induced Fas-dependent lymphocyte apoptosis, as Vlahakis et al. demonstrated R5 envelope activation of caspase-8. 16,17 Thus, published data support a proapoptotic role for CCR5, and both proapoptotic and antiapoptotic functions for CXCR4, depending on the in vitro system employed.
We first asked whether long-term NHL patient-derived BLCLs would conform to the pattern of CXCR4 overexpression described for tumors of neural and other tissue origin. Flow cytometric analysis of surface CXCR4 and CCR5 using monoclonal antibodies (mAbs) 12G5 and 2D7, respectively (R&D, MN), revealed that four of six EBV+ and two of three EBV− lines strongly expressed surface CXCR4 (Fig. 1). In contrast, most BLCLs failed to express high surface CCR5, consistent with published mRNA studies. 21,22 Confirmation of an apparent CXCR4 dependency for patient-derived BLCLs raised the question of differential impacts of CXCR4 vs. CCR5 signaling in the context of normal B cell transformation by EBV. In particular, we were interested in the effects of X4 gp120 engagement of CXCR4 vs. R5 gp120 engagement of CCR5 in the context of EBV-mediated B cell transformation.

Overexpression of surface CXCR4 vs. CCR5 on long-term B cell lymphoblast cell lines. CXCR4 exceeded CCR5 surface expression on all EBV+ BLCLs tested, with strongly positive cells exceeding 30% in four of six EBV+ lines. Two of three EBV− lines tested were strongly positive for CXCR4, with minimal CCR5 expression. The one BLCL (#2289) with CCR5 > CXCR4 expression had relatively low levels of both CKRs.
To gain insight into possible roles of gp120-induced CKR signal transduction in B cell lymphomagenesis, we assessed the impact of X4 vs. R5 gp120 on in vitro EBV transformation of normal B cells, defined by colony outgrowth in the fourth week (day 23). At this time point, no detectable replicating cells remained in control wells unexposed to EBV, indicating undetectable levels of endogenous EBV-induced transformation under our experimental conditions. Normal adult donor PBMCs, depleted of T cells by two rounds of immunomagnetic α-CD2 bead-negative selection (Dynal, Lake Success, NY) to eliminate EBV-specific T cells (<1% CD3+ by flow cytometry), were cultured in RPMI 1640 + 20% fetal calf serum (FCS) at 2 × 105 cells/200 μl in 96-well flat bottom plates. To increase the sensitivity for detection of enhancement, we used inefficiently transforming Type 2 AG876 EBV as well as the Type 1 B95.8 EBV usually employed for in vitro studies of EBV-induced transformation.
Replicate groups of 10 microwells were exposed to AG876 or B95.8 EBV in the presence or absence of 0.5 μg/ml gp120 from either R5 HIV-1 BaL or X4 HIV-1 IIIB, produced in the baculovirus SF9 cell system (Protein Sciences, Meriden, CT). In some wells 100 ng/ml SDF-1α and/or β (R&D) was added to EBV or EBV plus gp120-treated cells. Various controls received no EBV (10 wells), EBV in complete media only (20 replicates), or EBV plus SF9 cell extract subjected to the same purification steps as the envelope preparations (10 replicate wells). Positive controls received EBV plus α-CD40 mAb (0.1 μg/ml, BioSource, Camarillo, CA), a known costimulus for B cell transformation. Wells were microscopically inspected for blastoid colony clusters at 7, 14, and 21 days for the presence of expanded clones, and pulsed on day 23 with tritiated thymidine for 6 h to measure proliferation. Results of thymidine incorporation were analyzed by Kruskal–Wallis one-way ANOVA.
For donor “A,” both α-CD40 and X4 IIIB rgp120 added at the time of EBV exposure significantly enhanced proliferation 23 days later (p < 0.01, Fig. 2A). Five of five randomly chosen X4 gp120 wells were successfully expanded into long-term immortalized cell lines. Using another donor's cells, similar day 23 proliferative enhancement was detected whether IIIB gp120 was added with EBV, or up to 4 days later, whereas α-CD40 had only a modest effect (Fig. 2B). The R5 BaL envelope had no enhancing effect. Rather, in some experiments with a additional donors and Type 1 B 95.8 EBV, with high day 23 proliferation in the EBV exposed, untreated control cultures, BaL gp120 was actually inhibitory (Fig. 2C).
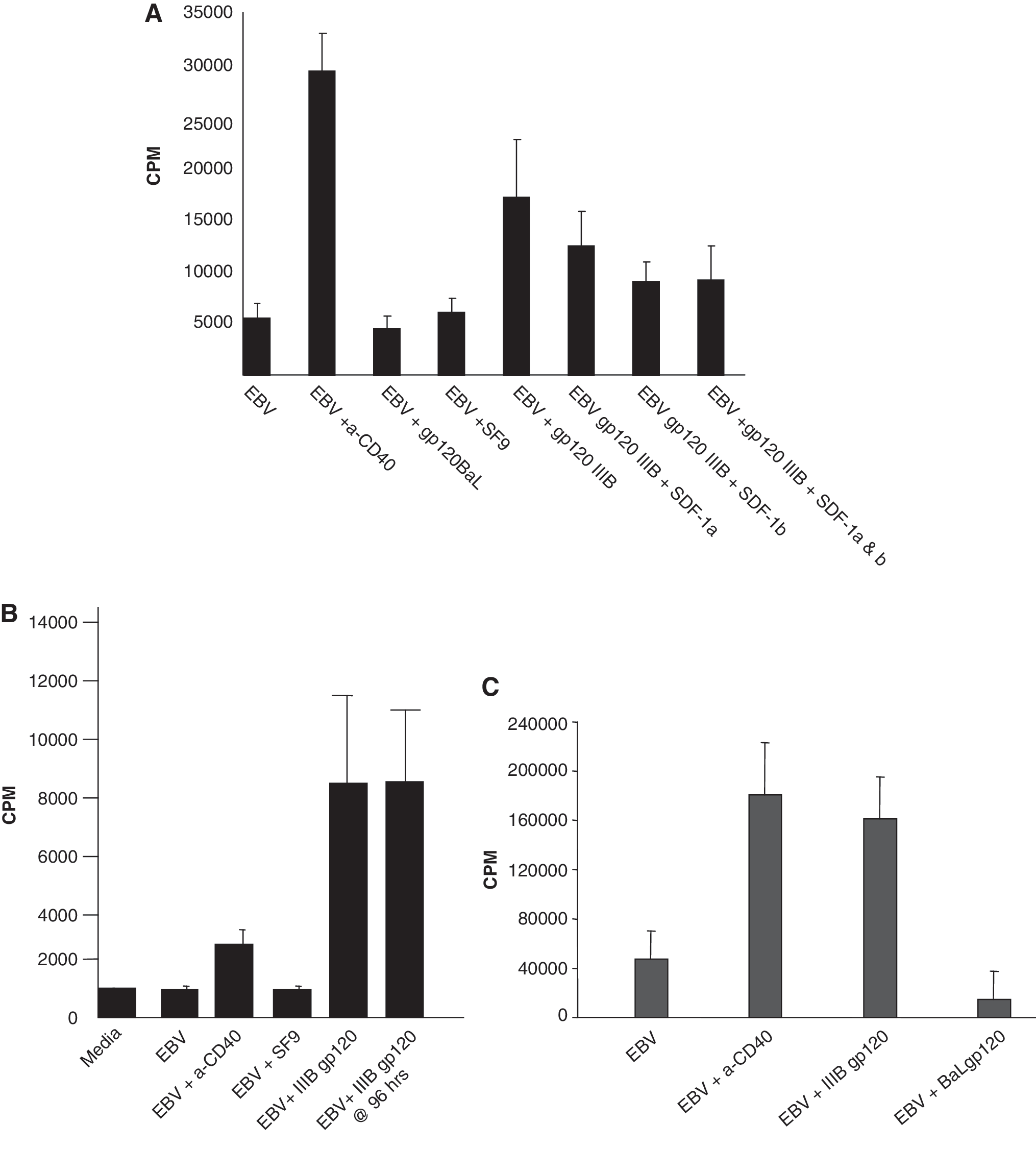
Enhancement of EBV-induced continuous B cell proliferation by X4 gp120 but not R5 gp120.
Neither SDF-1α nor SDF-1β enhanced proliferation induced by EBV (not shown) or EBV + IIIB gp120 (Fig. 2A). In fact, there was a significant reduction of gp120 enhancement when these CXCL12 species were present (p < 0.05). Differences between SDF-1 and gp120 in specific residue binding may account for differences in signal transduction and proliferative outcome. 23,24 CXCR4-specific 12G5 mAb also failed to enhance growth, but agonistic 12G5 mAb effects might not occur at the saturating concentrations used. Also, recent work 12 indicates that the CXCL12 receptor, CXCR7, is upregulated and crucial during EBV transformation. Absent cross-reactivity, 12G5 would not mimic any gp120 effects mediated by CXCR7 binding. Our use of normal B cells, rather than transformed gliomas, 21 established Type 1 lymphoma lines, 25 or in vivo-generated lymphomas 8 may explain why we also did not confirm previous reports of SDF-1 promoting growth. Indeed, it would have been surprising to find that a natural ligand for CXCR4 was tumorigenic for normal B cells in the presence of EBV.
B cell lymphomagenesis in the HIV+ setting is multifactorial. These experiments suggest that HIV CKR tropism may be one piece of the puzzle, and are consistent with the increased appearance of X4 virus and NHL in advanced disease. These are the first studies to implicate gp120-induced signaling as a factor in the earliest stages of normal cell transformation. Inefficient transformation by Type 2 EBV may help reveal X4 gp120-enhancing effects, whereas much more robust Type 1 transformation may provide the context necessary for detecting R5 inhibition (Fig. 2C). The relative contributions of gp120 signaling through CXCR4 vs. CXCR7 remain to be explored.
Footnotes
Acknowledgments
This work was funded in part by American Cancer Society Institutional Research Grant IRG11-36 to S.I. and NIH Grant N01-AI45207. We thank Dr. Richard Ambinder for the gift of patient-derived BLCLs. Both authors contributed equally to the design, performance, and interpretation of this work.
Author Disclosure Statement
No competing financial interests exist.