
Abstract
The HLA-B*27 allele is overrepresented in patients who control HIV-1 replication without antiretroviral therapy. CD8+ T cell responses that target the immunodominant KK10 epitope in Gag are thought to play a major role in this control, and escape at R264 of KK10 is often associated with dramatic virologic breakthrough. We present a case in which an HLA-B*27-positive chronic progressor transmitted HIV-1 to an HLA-B*27-positive viremic controller who was temporarily on HAART, but who has since controlled viremia for over 4 years. We hypothesized that differences in the KK10 epitope of these patients would affect pathogenesis and viral fitness, but found no correlation between autologous KK10 mutations and disease progression or between the predicted fitness impact of autologous HLA-B*27-associated mutations and the actual fitness of autologous virus. This case of viral transmission between two HLA-B*27-positive individuals provides further evidence that prolonged control of fully pathogenic HIV-1 is possible.
A
One of the factors in the control of viremia is the cytotoxic T lymphocyte (CTL) response. A polyfunctional, proliferative CTL response to HIV antigens, and specifically to the Gag polypeptide, is associated with control. 1 Additionally, the HLA alleles B*57 and B*27 are overrepresented in patients who effectively control viremia. These alleles predominantly present peptides from Gag, and the HLA-B*27 response is dominated by the presentation of a single epitope in Gag, KK10. 3 –5 CTL responding to KK10 are polyfunctional and have superior functional avidity. 6
Here we document transmission of HIV-1 between two HLA-B*27 individuals who present with dramatically different disease progressions. The progressor (chronic progressor, CP) transmitted to an HLA-B*27 individual who was on highly active antiretroviral therapy (HAART) for 4 years, but has since maintained considerable control of the virus for over 4 years since terminating treatment (VC). The progressor was diagnosed in 1994; the VC was diagnosed in 1997 but may have been infected anytime between 1994 and 1997. We observed no drug resistance mutations in RT and protease of the VC as assessed by the Stanford HIV Drug Resistance Database (GenBank accession numbers: HM208365 and HM208366). The VC consistently maintains CD4 counts above 500 cell/μl and a viral load of less than 2000 copies/ml (Fig. 1). It has been shown that structured treatment interruption (STI) of HAART during acute infection can result in transient control of HIV-1 viremia after treatment cessation, with 7 and without 8 the addition of interleukin (IL)-2. The majority of these patients controlled viral replication for less than 2 years, however. 9 This patient has therefore maintained remarkable control of viremia since terminating HAART.

Viral load and CD4 T cell count for (
The HLA-B*27-restricted KK10 epitope in Gag is immunodominant in most HLA-B*27 individuals, and escape via an arginine-to-lysine substitution at residue 264 of Gag correlates with loss of control of viremia. 4,10,11 This mutation has a high fitness cost and is associated with a compensatory S173A substitution upstream in Gag. “Fitness” refers to the capacity of the virus to infect and replicate in activated CD4+ T cells. 12 We hypothesized that differences in the mutations at KK10 would correlate with the different disease progression of these patients.
Using reverse transcriptase nested polymerase chain reaction (PCR) we amplified gag and significant portions of the rest of the HIV-1 genome from both VC and CP. We observed a six-amino acid insertion in p6 of Gag in both patients' sequences (Fig. 2), which, interestingly, has been associated with a decreased virologic response in patients receiving unboosted amprenavir. 13 Considering that both patients possessed the insertion, it is unlikely to have played a role in the different disease progression of the individuals, but it did serve as evidence of transmission. To confirm transmission, we constructed a neighbor-joining tree consisting of plasma and proviral sequences from both patients, as well as the B clade sequences found to be most closely related to the patient samples by the HIV BLAST program from the Los Alamos National Laboratory website (GenBank accession numbers: HM208361–HM208364) (Fig. 3). Sequences from the patients grouped together, supporting the assertion that transmission occurred between the two individuals.

Sequence of VC and CP proviral and plasma Gag aligned with reference strain HXB2. The HLA-B*27 KK10 epitope is highlighted, as is the putative upstream compensatory mutation site, S173. The 6 amino acid insertion common to these patients is also highlighted.
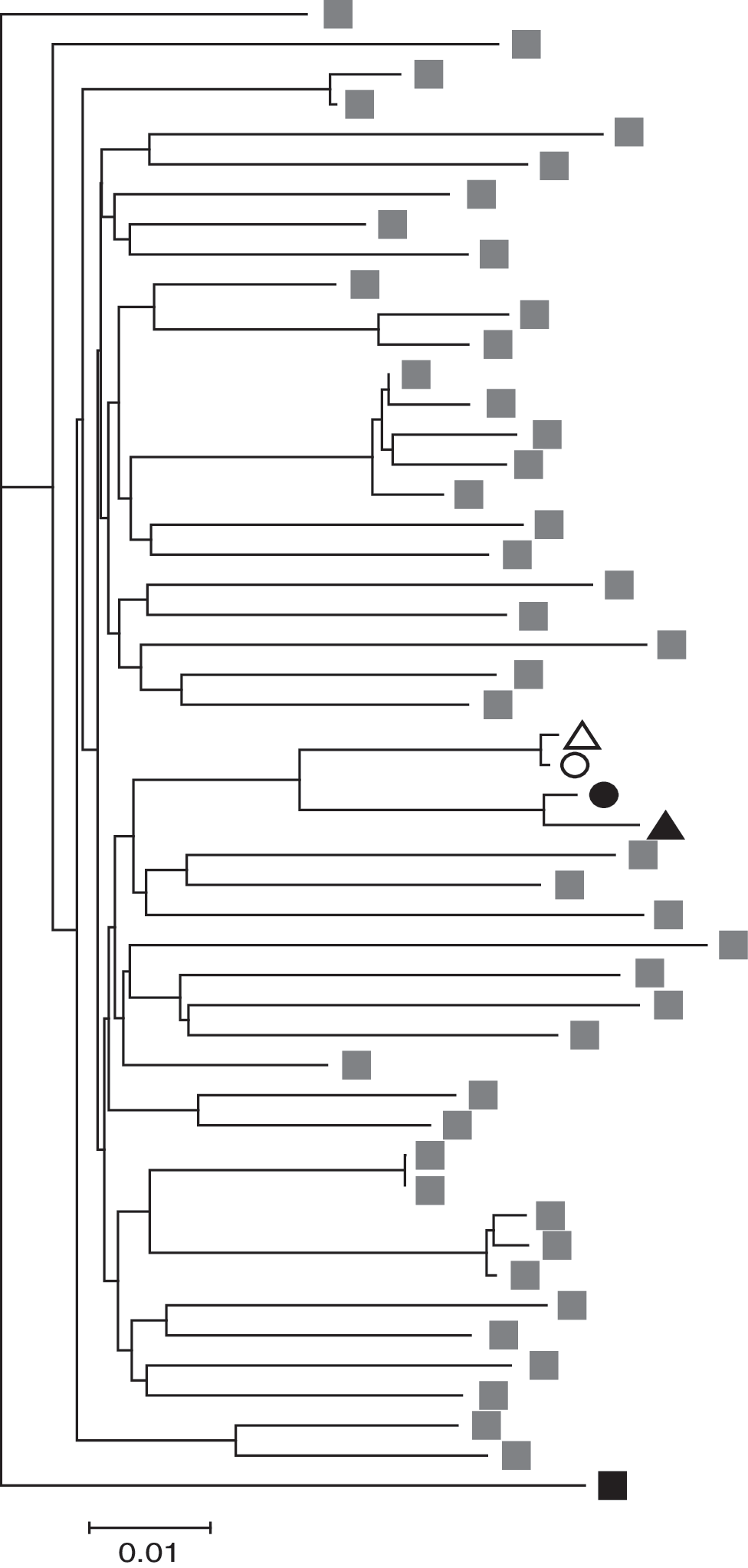
Phylogenetic analysis of plasma (triangle) and proviral (circle) sequences from VC (open symbols) and CP (black symbols). The LANL HIV BLAST program was used to identify the B Clade sequences that are most closely related to the patients' sequences; these are indicated with gray symbols. Black symbol represents a D Clade outgroup sequence.
Both patients expressed the L268M mutation in the KK10 epitope, but maintained different mutations at the R264 residue. The L268M mutation is a common early escape mutation in KK10 but is of limited importance as an effective, de novo CTL response to this variant usually develops. 14 At R264, the progressor possessed the common R264K escape mutant but lacked the putative compensatory mutation at S173; the viremic controller, in contrast, had a rare R264Q substitution (Fig. 2). It is unclear whether the R264Q mutant in the VC developed from wild-type virus or from virus that already possessed the R264K mutation. Considering, however, that the codons for R264, R264K, and R264Q are AGA, AAA, and CAA, respectively, it seems likely that R264Q developed from a transmitted isolate that contained the R264K residue. In vitro work has shown that the R264Q mutation has less of a fitness cost than R264K, but still provides effective escape from HLA-B*27-specific CTL. 15,16 Thus based on this combination of mutations, we anticipated that the Gag of the viremic controller would be more fit than that of the progressor.
To directly assess differences in the fitness of Gag from these patients, we amplified gag from both patients and inserted the gene into an NL43 pseudotype virus that has GFP inserted into the env gene. We then used a CXCR4 env plasmid to make virus capable of a single round of infection, and compared the infectivity of the constructs by using GFP to measure infection. As shown in Fig. 4A, the virus containing gag derived from the progressor was, surprisingly, more fit than that derived from the viremic controller.
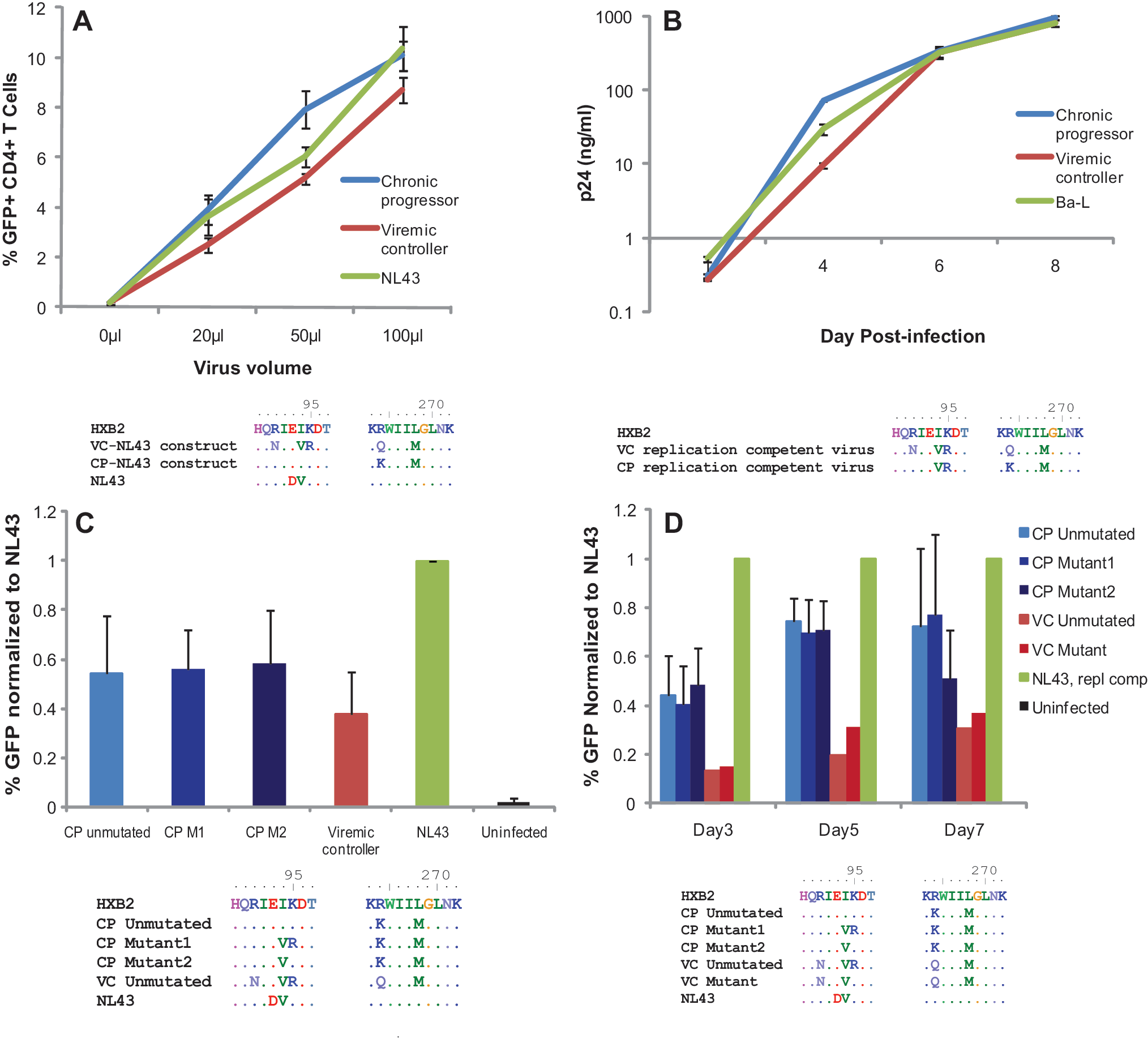
Fitness of CP and VC virus and viral constructs with sequences of relevant gag regions displayed below each experiment. (
To assess the relative impact of these mutations on whole viral fitness, we also compared the fitness of replication-competent virus derived from the two patients. Isolates from the VC and CP were cultured from CD4 T cells and plasma virus, respectively. Figure 4B shows that these replication-competent viruses had similar fitness both to each other and to the Ba-L laboratory strain. Interestingly, the replication-competent virus from the progressor had developed a two amino acid substitution, I94V and K95R, in p17 of Gag, however, which was not seen in other sequences from the progressor but was present in clones amplified from the viremic controller. We examined the database of B clade sequences on the Los Alamos website and found that these mutations are not uncommon in the B clade. Nonetheless we hypothesized that these mutations arose during the culturing of replication-competent virus from the progressor because they served as compensatory mutations for the R264K mutation in this patient.
To examine this possibility, we used site-directed mutagenesis to insert these substitutions into a CP-derived gag clone (CP mutant 1). Additionally, we generated another Gag clone from the CP in which we mutated just residue I94 to V(CP mutant 2), as this is the consensus residue for B clade HIV-1. Figure 4 illustrates the sequences tested in the fitness assay. We inserted these gag mutants into the NL43 pseudotype virus as previously described, but found little difference between the three CP mutant clones. However, all CP mutant clones were more fit than the VC-NL43 construct (Fig. 4C).
Because these mutations were in gag, we were concerned that their effects might not be proprerly characterized in a single round of infection. Single-cycle infection reflects fitness differences that affect viral lifecycle events up until the integration of HIV-1 DNA into the host genome and expression of that genome. However, it may fail to distinguish mutations that affect virion formation or release from the cell. To address this concern, we inserted our gag mutant clones into an NL43 replication-competent backbone. This vector has GFP inserted into nef, but has an intact env gene. We also used site-directed mutatgenesis to mutate gag from the VC such that it too possessed the B clade consensus residues 94V and 95K. Over the course of 7 days, we found no difference in the fitness of the CP mutants, and substitutions in residues 94 and 95 in VC also had little effect (Fig. 4D).
Having thoroughly examined the relative fitness of virus from these patients, we conclude that the CP had slightly more fit virus than the VC in vitro. This finding is in contrast to what was predicted based on in vitro studies that evaluated the fitness impact of KK10 mutants.
The VC in this study maintained remarkable control of a fully pathogenic virus following cessation of HAART despite acquiring an effective escape mutation in the KK10 epitope. This is remarkable, as escape at the KK10 epitope generally occurs late in infection and is associated with an increase in viremia. The specific R264Q/L268M mutation combination present in the VC, in fact, was associated with a dramatic increase in viremia and low CD4 T cell counts in the first, and one of the only, published descriptions of this mutation. 16
Although limited sample availability has not permitted us to perform more thorough immunologic analyses of these patients, it is interesting to speculate that HAART reset the viral load to a level that this patient could then control via her immune response. Additionally, this brief period of treatment may have affected how the virus mutated in response to the immune response, perhaps epitomized by the development of an uncommon variant at KK10. Understanding the kinetics of viral escape mutation at low and high levels of viremia may help us define the immune mechanisms responsible for the spectrum of HIV-1 progression. This would greatly improve our ability to predict the pathogenesis of infection in infected individuals.
Footnotes
Acknowledgment
This work was supported by NIH Grant R01 AI080328 (J.N.B.).
Author Disclosure Statement
No competing financial interests exist.