
Abstract
To gain a better understanding of the assembly process in simian immunodeficiency virus (SIV), we first established the conditions under which recombinant SIV Gag lacking the C-terminal p6 domain (SIV GagΔp6) assembled in vitro into spherical particles. Based on the full multimerization capacity of SIV GagΔp6, and to identify the Gag sequences involved in homotypic interactions, we next developed a pull-down assay in which a panel of histidine-tagged SIV Gag truncation mutants was tested for its ability to associate in vitro with GST-SIVGagΔp6. Removal of the nucleocapsid (NC) domain from Gag impaired its ability to interact with GST-SIVGagΔp6. However, this Gag mutant consisting of the matrix (MA) and capsid (CA) domains still retained 50% of the wild-type binding activity. Truncation of SIV Gag from its N-terminus yielded markedly different results. The Gag region consisting of the CA and NC was significantly more efficient than wild-type Gag at interacting in vitro with GST-SIVGagΔp6. Notably, a small Gag subdomain containing the C-terminal third of the CA and the entire NC not only bound to GST-SIVGagΔp6 in vitro at wild-type levels, but also associated in vivo with full-length Gag and was recruited into extracellular particles. Interestingly, when the mature Gag products were analyzed, the MA and NC interacted with GST-SIVGagΔp6 with efficiencies representing 20% and 40%, respectively, of the wild-type value, whereas the CA failed to bind to GST-SIVGagΔp6, despite being capable of self-associating into multimeric complexes.
Introduction
T
During assembly, Gag is cleaved by the virus-encoded protease to generate the functional proteins of the mature virion. Processing of the simian immunodeficiency virus (SIV) Gag precursor generates the mature proteins: matrix (MA), which lines the inner side of the lipid viral envelope, capsid (CA), which forms the characteristic cone-shaped shell of the viral core, nucleocapsid (NC), which is complexed to the genomic RNA within the viral core, and p6, which is implicated in virion budding and release. 6,7 In addition, two spacer peptides are generated by proteolytic cleavage of SIV Gag: SP1 and SP2, which separate the CA and NC and the NC and p6 domains, respectively. 7 We have demonstrated that the MA domain of the SIV Gag precursor plays crucial roles during viral assembly: it not only provides, through an N-terminal myristoyl moiety and a polybasic region comprising residues 26 to 32, the determinants necessary for the proper targeting and association of Gag with the plasma membrane, 8,9 but is also involved in Env incorporation into virions through its specific interaction with the Env cytoplasmic domain. 10 –12 For human immunodeficiency virus type 1 (HIV-1), biochemical and structural analyses have shown that Gag membrane binding is regulated by a myristoyl switch mechanism whereby Gag multimerization and binding of MA to phosphatidylinositol-(4,5)-biphosphate [PIP(4,5)P2] trigger the exposure of the myristate group. 13,14 Interestingly, it has recently been proposed that RNA binding to the HIV-1 MA polybasic region prevents premature nonspecific binding of Gag to cellular membranes prior to its association with the PIP(4,5)P2-containing plasma membrane. 15,16
Several studies in HIV-1 indicate that the CA and NC domains of Gag are important for Gag-Gag interactions. The CA protein consists of an N-terminal domain (NTD) and a C-terminal domain (CTD) connected by a flexible linker. Electron microscopy reconstruction analysis together with X-ray crystallography studies have recently shown that the structure of the HIV-1 CA lattice in the mature core is formed from hexamers of the NTD linked by CTD dimers. 17,18 These structural data help explain why mutations within both the CA NTD and CTD are detrimental to HIV-1 particle production. 19 –21 In addition, accumulating data indicate that the HIV-1 NC domain also mediates Gag multimerization, which appears to be related to its intrinsic ability to bind RNA. 22 –25
Although SIV is closely related to HIV-1, their Gag polyproteins display certain differential biological features. Indeed, the SIV CA domain does not bind cyclophilin A, whereas in HIV-1 the CA–cyclophilin A association is necessary for the particle uncoating step that follows virus entry. 26,27 Furthermore, it has been shown that the relative contribution of the NC zinc-finger domains to genomic RNA binding is different in SIV and HIV-1. 28 Moreover, analysis of the determinants in the p6 domain of Gag that are required for the packaging of the accessory protein Vpr has revealed significant differences between HIV-1 and SIV 29 : in HIV-1, a dileucine motif present at the p6 C-terminus is responsible for Vpr incorporation into virions. In contrast, the virion association motif for SIV Vpr is located at the N-terminal half of p6. The latter motif, which is absent from HIV-1 Gag, also mediates the packaging of Vpx into SIV particles. Interestingly, we have demonstrated that the SIV MA, in contrast to its HIV-1 counterpart, has the ability to self-assemble into lentivirus-like particles. 8,30 The release of VLPs formed by the SIV MA alone may be promoted by the presence at the C-terminus of this viral protein of a redundant PTAP motif, which is characteristic of functional retroviral late-budding domains.
Given these differential features of the SIV Gag precursor, the study of the determinants in this protein that drive SIV particle assembly is relevant, not only to clearly establish the similarities and differences of the assembly process among retroviruses, but also to gain a better understanding of lentivirus morphogenesis. Taking this into account, we therefore decided to map the sequences in SIV Gag that are involved in protein–protein interactions and Gag multimerization. We developed an in vitro binding assay in which we examined the ability of a panel of SIV Gag subdomains to interact with the Gag precursor. These experiments, together with VLP assembly studies performed in cell culture, allowed us to determine the contribution of each SIV Gag domain to Gag–Gag interaction and to define the minimal SIV Gag subdomain that interacts with Gag in vitro as efficiently as the full-length precursor.
Materials and Methods
Plasmid constructs
The gag-coding sequences for all the expression plasmids were derived from the proviral clone SIVSMM PBj1.9. 8 SIV Gag lacking the C-terminal p6 domain (SIV GagΔp6; PBj1.9 Gag residues 1–448) was expressed both as fusion with Schistosoma japonicum glutathione S-transferase (GST) and with an N-terminal histidine tag by cloning the corresponding gag open reading frame into the pGEX-2T (GE Life Sciences) and the pET-30b(+) (Novagen) plasmid vectors, respectively. All the gag-derived constructs were cloned into the pET-30b(+) vector to express in Escherichia coli the His-tagged SIV Gag subdomains used in the pull-down assays. For expression in mammalian cells, the entire SIV gag gene [nucleotides (nt) 829 to 2352 of the SIVSMM PBj1.9 genome] was cloned into the KpnI and EcoRI sites of pCDNA3.1(+) (Invitrogen).
The gag genes carrying internal deletions within the CA-coding region were generated by substituting the mutated PflMI-PflMI restriction fragment (nt 1214–2101) for the wild-type counterpart in the pcDNA-SIVgag plasmid. For expression of His-tagged Gag1–448 (GagΔp6), Gag136–365 (CA), and Gag287–448 in mammalian cells, the His-coding expression cassette together with the gag sequences were excised from the corresponding pET plasmids and subcloned into the NheI and NotI sites of the pcDNA3.1(+) vector. The sequences of primers and details of the cloning strategies are available on request.
Expression in E. coli and purification of recombinant proteins
Expression in E. coli BL21 (DE3) and purification of recombinant proteins followed procedures described previously. 12,31 Bacterial extracts for the purification of the recombinant GST-SIVGagΔp6 protein were obtained by sonication in phosphate-buffered saline (PBS) containing 0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) and protease inhibitor cocktail (Roche Applied Science). After treating the extracts with DNase I, insoluble material was removed by centrifugation for 10 min at 16,000 × g. The supernatants were mixed with 50% (w/v) slurry glutathione-Sepharose 4B (GE Life Sciences) and incubated for 30 min at room temperature. Beads were then washed three times with 10 bed volumes PBS. Recombinant histidine-tagged Gag-derived proteins were purified by immobilized metal ion adsorption chromatography (His Microspin Purification Module, GE Life Sciences) essentially as we have recently described. 12 Protein concentrations were estimated as described previously. 12 For the in vitro assembly reactions (see next section), an aliquot of protein extracts was further treated with DNase I (5 units; Promega) and RNase A (50 μg/ml; Sigma-Aldrich) to remove nucleic acids before purification by affinity chromatography. Potential contamination of purified Gag proteins with nucleic acids was assessed by both spectrophotometric determination of the A 260/A 280 ratio and agarose gel electrophoresis after phenol extraction and ethanol precipitation of protein samples.
In vitro assembly of SIV GagΔp6
Purified His-tagged SIV GagΔp6 stored at −80°C was thawed on ice and centrifuged at 16,000 × g for 20 min. Aliquots from the supernatants were then used in the assembly reactions. Five microrams of recombinant Gag protein was incubated for 3 h at 37°C in 25-μl reactions containing 50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 5 mM dithiothreitol (DTT), 10 μM ZnCl2, 500 ng of in vitro transcribed R-U5-MA viral RNA (see next section), and 100 units of recombinant RNasin ribonuclease inhibitor (Promega). Assembly reactions were analyzed by both sedimentation assays 32 and electron microscopy. Particles formed under the conditions described above were collected by centrifugation for 1 h in an Eppendorf microcentrifuge at 16,000 × g at 4°C. Supernatant and pellet fractions were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and proteins blotted onto nitrocellulose membranes. Detection of His-tagged proteins in the pellet and supernatant fractions was performed by Western blotting using a horseradish peroxidase (HRP)-conjugated antibody specific for penta-His (QIAGEN). Western blots were developed with an enhanced chemiluminescence and chemifluorescence assay (ECL Plus Reagent, GE Life Sciences).
For the electron microscopy analyses, the assembly reactions were applied onto Formvar-coated grids (TAAB Laboratories, UK) and negatively stained with 2% uranyl acetate. Grids were visualized using a Jeol transmission electron microscope operating a 80 kV. In the case of the His-CA and His-CAΔCTD (Gag136-274) proteins, the in vitro assembly reactions were performed at a protein concentration of 1 μg/μl in buffer (pH 8.0) containing 1 M NaCl, and the assembly products were analyzed by SDS–PAGE. The CA-derived assemblies were further examined by electrophoresis on nondenaturing 5% polyacrylamide gels. The molecular mass of the SIV CA-related oligomeric complexes was estimated based on the relative mobilities of the molecular weight protein standards for native electrophoresis (High Molecular Weight Calibration kit for native electrophoresis, GE Life Sciences).
In vitro transcription
The sequence spanning nt 293–1218 of the SIVSMM PBj1.9 genome, which corresponds to the R and U5 regions of the 5′ long terminal repeat together with the first 390 nt of the gag gene (referred to here as the R-U5-MA construct), was cloned into the NcoI–EcoRV sites of the pGEM-5Zf plasmid (Promega) and used as template for RNA synthesis essentially as we have previously described. 31 For the production of large amounts of in vitro-transcribed R-U5-MA RNA we used the RiboMAX Large Scale RNA Production System (Promega). Briefly, RNA was synthesized in a final volume of 20 μl containing 500 ng of SalI-linearized plasmid; 80 mM HEPES-KOH (pH 7.5); 24 mM MgCl2; 2 mM spermidine; 40 mM DTT; 7.5 mM (each) ATP, GTP, CTP, and UTP; and 2 μl of the T7 enzyme mix (mixture of T7 RNA polymerase, RNasin ribonuclease inhibitor, and yeast inorganic pyrophosphatase). The reaction mixture was incubated 2 h at 37°C followed by treatment with 2 units of RNase-free DNase I (Promega) and further incubated at 37°C for 15 min to remove the DNA template. The reaction was extracted twice with a 1:1 mixture of phenol and chloroform and then precipitated in ethanol. The RNA product was resuspended in water and stored at −80°C until further use.
In vitro binding assays for Gag–Gag interaction
Five micrograms of GST-SIVGagΔp6 or GST proteins was prebound to glutathione-Sepharose 4B and incubated with 1 μg purified His-tagged wild-type or mutant SIV Gag proteins in 100 μl PBS containing 0.5% CHAPS, 1% bovine serum albumin (BSA), and protease inhibitor cocktail for 3 h at 4°C. CHAPS was chosen because it is a zwitterionic detergent that prevents protein aggregation while preserving protein–protein interactions. The resin-bound proteins were washed six times with 40 bed volumes of binding buffer. The protein complexes were eluted from the glutathione resin by resuspending the beads in Laemmli sample buffer, resolved by SDS–PAGE, and detected by Western blotting using the HRP-conjugated anti-His antibody. The levels of protein recovery for each His-tagged Gag subdomain were determined by comparison to known amounts of the corresponding purified recombinant protein by Western blotting and referring the data to those obtained with wild-type Gag, considered as 100%. Quantitation of Western blot signals was performed as previously described. 11
ELISA-based protein interaction assay
Six micrograms of purified GST-SIVGagΔp6 fusion protein or GST was bound to glutathione-coated wells (Reacti-Bind Glutathione Coated plates, Pierce) for 1 h at room temperature in binding buffer (PBS–0.5% CHAPS), and then washed three times with 200 μl binding buffer. Dilutions of His-SIVGagΔp6 or His-tagged Gag subdomains in 100 μl binding buffer containing 0.1% BSA were incubated with immobilized GST-SIVGagΔp6 or GST for 16 h at 4 °C. Wells were washed with binding buffer and protein complex formation was detected by the addition of HRP-conjugated anti-His antibody and 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) substrate. The resulting colored reaction signal was measured on a microtiter plate (enzyme-linked immunosorbent assay, ELISA) reader at 405 nm (reference wavelength 490) as previously described. 11 Gag binding was expressed as the amount (in arbitrary units) of His-SIVGagΔp6 immunoreactivity found associated with GST-SIVGagΔp6 minus that associated with GST alone.
Cells and viruses
COS-7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM; HyClone) supplemented with 10% fetal bovine serum (GIBCO Cell Culture Systems, Invitrogen). The recombinant vaccinia virus vTF7-3 expressing the T7 RNA polymerase was kindly provided by Dr. B. Moss (NIH, MD).
Analysis of protein expression in mammalian cells
The expression of wild-type Gag, His-tagged Gag subdomains, or CA deletion mutants in mammalian cells was carried out using the vaccinia virus T7 system 33 since the gag constructs were cloned into the pCDNA3.1(+) plasmid that carries the T7 RNA polymerase promoter. Confluent monolayers of COS-7 cells (35-mm-diameter dishes) were infected with the vTF7-3 recombinant vaccinia virus at a multiplicity of infection of 10 for 1 h at 37°C. After infection, the cells were washed twice with DMEM, and then transfected with the plasmid constructs using GeneJammer Transfection Reagent (Stratagene). In the case of the coexpression of wild-type SIV Gag with mutant His-Gag287–448, initial experiments showed that cotransfection of the corresponding plasmids at a 1:1 mass ratio resulted in intracellular levels of His-Gag287–448 significantly higher (at least 20-fold) than those of wild-type SIV Gag. Therefore, to attain comparable intracellular levels of wild-type Gag and mutant His-Gag287–448 proteins, plasmid DNAs were cotransfected at both 20:1 and 10:1 mass ratios in the definitive experiments shown in Fig. 6.
Thirty hours posttransfection, cells were harvested and lysed at 4 °C in lysis buffer [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate] containing protease inhibitor cocktail. The culture supernatants from the Gag-expressing cells were filtered through 0.45-μm-pore-size syringe filters and VLPs were pelleted from the clarified supernatants by ultracentrifugation (100,000 × g, 90 min, 4°C) through a 20% (w/v) sucrose cushion essentially as we have previously described. 11 Cell- and VLP-associated proteins were resolved on SDS-polyacrylamide gels, blotted onto nitrocellulose membranes, and analyzed by Western blotting as described above. His-tagged SIV Gag-derived proteins were detected using the anti-His antibody or the SIV CA-specific KK60 monoclonal antibody. The CA deletion mutants were detected by using the MA-specific KK59 monoclonal antibody. 12
Results
In vitro assembly properties of SIV Gag
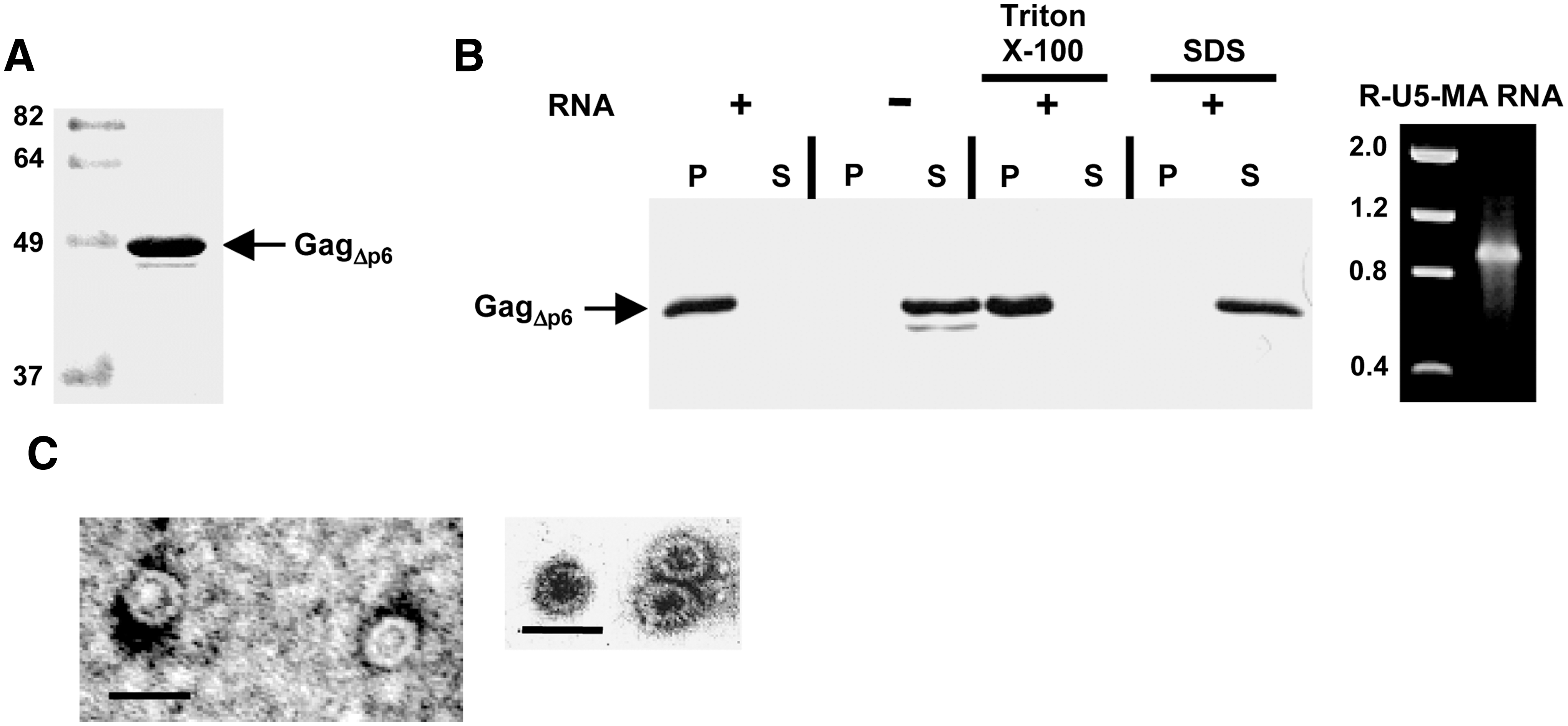
The SIV Gag precursor lacking the p6 domain (SIV GagΔp6) was expressed in E. coli either as fusion with GST or carrying an N-terminal histidine tag. We decided to utilize in our assays these recombinant SIV GagΔp6 proteins because they could be readily purified from E. coli as soluble proteins, whereas their full-length counterparts were unstable and extensively degraded. A similar instability behavior in E. coli has consistently been observed for full-length HIV-1 Gag. 22,34 As shown in Fig. 1A, purification of His-SIVGagΔp6 by affinity chromatography resulted in protein preparations that were more than 90% pure as judged by SDS–PAGE.
In vitro assembly of SIV GagΔp6.
We first evaluated whether His-SIVGagΔp6 had the ability to self-assemble in vitro. To this end, the purified protein was incubated at 37°C for 3 h under the conditions described in Materials and Methods, and the assembly reaction was then centrifuged to separate the pelletable assembled products from the unassembled Gag molecules using a sedimentation assay similar to that employed for the analysis of the in vitro assembly of HIV-1 Gag. 32 The analysis of the resulting fractions by Western blotting showed that His-SIVGagΔp6 partitioned with the pellet fraction when an in vitro-transcribed RNA consisting of the SIV R, U5, and MA genomic regions was added to the assembly mixture (Fig. 1B). In contrast, the purified Gag protein was recovered in the supernatant fraction when the RNA was omitted from the assembly reaction (Fig. 1B). Moreover, the assembly of His-SIVGagΔp6 was not affected by the presence of the nonionic detergent Triton X-100 [0.5% (v/v)]. As expected, addition of SDS to a final concentration of 0.1% (w/v) completely blocked the formation of assembled products (Fig. 1B).
Examination of the assemblies by negative-stain electron microscopy revealed that His-SIVGagΔp6 formed spherical particles whose diameter was approximately 35 nm and that were similar in appearance to immature virions without a membrane (Fig. 1C). These spherical structures were also observed when the protein samples were not treated with RNase A during their purification (data not shown), indicating that the bacterial RNA that copurifies in the protein preparations is sufficient to promote the in vitro assembly of SIV Gag. Overall, these results demonstrate that recombinant SIV GagΔp6 is able to multimerize in vitro into spherical particles.
Construction of His-tagged SIV Gag deletion mutants
Taking into account that SIV Gag lacking the p6 domain was capable of self-assembling in vitro, we decided to use this recombinant protein to map the domains in Gag involved in protein self-interactions. To this end, we generated a panel of deletion mutants in which SIV GagΔp6 (Gag1–448) was progressively truncated either from its C-terminus (mutants Gag1–393 to Gag1–231) or from its N-terminus (mutants Gag86–448 to Gag287–448) (Fig. 2). We also constructed plasmids encoding the mature SIV Gag proteins MA (Gag1–135), CA (Gag136–365), and NC (Gag383–448) (Fig. 2). All these SIV gag constructs were expressed in E. coli as His-tagged proteins and tested for their ability to interact with GST-SIVGagΔp6 in the in vitro binding assays described below.

Construction of SIV Gag truncation mutants. Schematic diagram showing the protein domains of the SIV Gag precursor and of the Gag mutants that were expressed in bacteria. The major homology region (MHR) in the CA as well as the N-terminal (ZFN) and C-terminal (ZFC) zinc-finger motifs in the NC are indicated. The names of the mutants refer to the SIV Gag amino acid positions, with 1 being the initiator methionine in the polyprotein.
In vitro binding assay for SIV Gag–Gag interaction
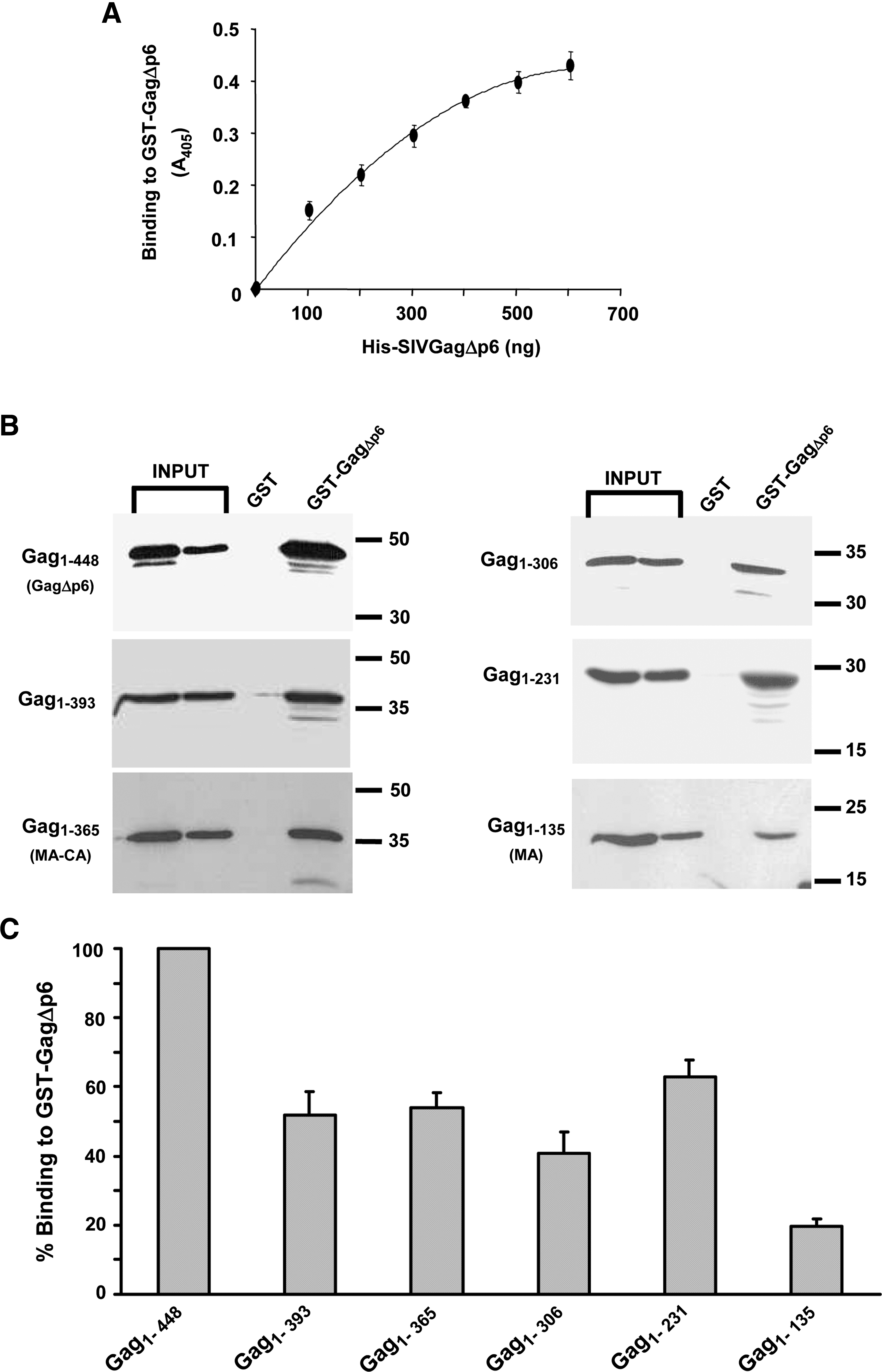
To study SIV Gag–Gag interaction, we adapted a GST pull-down assay that recently allowed us to demonstrate a physical interaction between the SIV MA protein and the Env cytoplasmic domain. 12 We first examined the ability of purified His-SIVGagΔp6 (Gag1-448) to interact with GST-SIVGagΔp6 immobilized onto a glutathione-Sepharose resin. The protein bound to GST-SIVGagΔp6 was then visualized by Western blotting using an anti-His antibody. As shown in Fig. 3B (panel for Gag1–448), His-SIVGagΔp6 associated in a specific manner with the GST-SIVGagΔp6 fusion protein, but not with GST alone, indicating that these recombinant Gag proteins are able to interact in vitro. Estimation of the amount of His-tagged Gag1–448 protein recovered on the beads (see Materials and Methods) showed that under the experimental conditions used in our pull-down assays, 40% of the total His-Gag1–448 binds to immobilized GST-SIVGagΔp6. Furthermore, analysis of the in vitro association between these recombinant Gag proteins by means of an ELISA-based binding assay indicated that the interaction of His-SIVGagΔp6 with GST-SIVGagΔp6 is saturable and is dose dependent (Fig. 3A).
Binding of His-tagged SIV Gag C-terminal truncation mutants to GST-SIVGagΔp6.
Effect of progressive truncation of the SIV Gag C-terminus on Gag–Gag interaction
We first examined whether progressive deletions from the carboxyl terminus of His-Gag1–448 had any effect on its ability to interact with GST-SIVGagΔp6 (Fig. 3B). The signal intensities on the Western blots from the pull-down assays were quantitated to estimate the binding efficiency to GST-SIVGagΔp6 of each mutant Gag protein with respect to that of wild-type Gag (His-Gag1–448) (see Materials and Methods). Deletion of the C-terminal 55 amino acids spanning the two NC zinc-finger motifs (Gag1–393) was sufficient to reduce the Gag–Gag in vitro interaction to approximately 50% of that of wild-type SIV Gag (Fig. 3B and C). Further deletions from the Gag C-terminus had no additional inhibitory effect on Gag interaction. Indeed, mutants Gag1–365 and Gag1–306 associated with GST-SIVGagΔp6 with efficiencies comparable to that of mutant Gag1–393. These results indicate that deletion of the NC from SIV Gag is detrimental to Gag self-association. However, the presence in the Gag constructs of the entire CA or of a CA-derived region allows substantial Gag–Gag interaction. In this regard, while the MA protein (Gag1–135) was still able to associate with GST-SIVGagΔp6 (Fig. 3B), albeit displaying only a 20% binding efficiency of that of wild-type Gag (Fig. 3C), the SIV Gag subdomain comprising the MA protein and the first N-terminal 96 residues of CA exhibited a binding capacity representing 62.9 ± 5.0 % of that of wild-type Gag.
Effect of progressive truncation of the SIV Gag N-terminus on Gag–Gag interaction
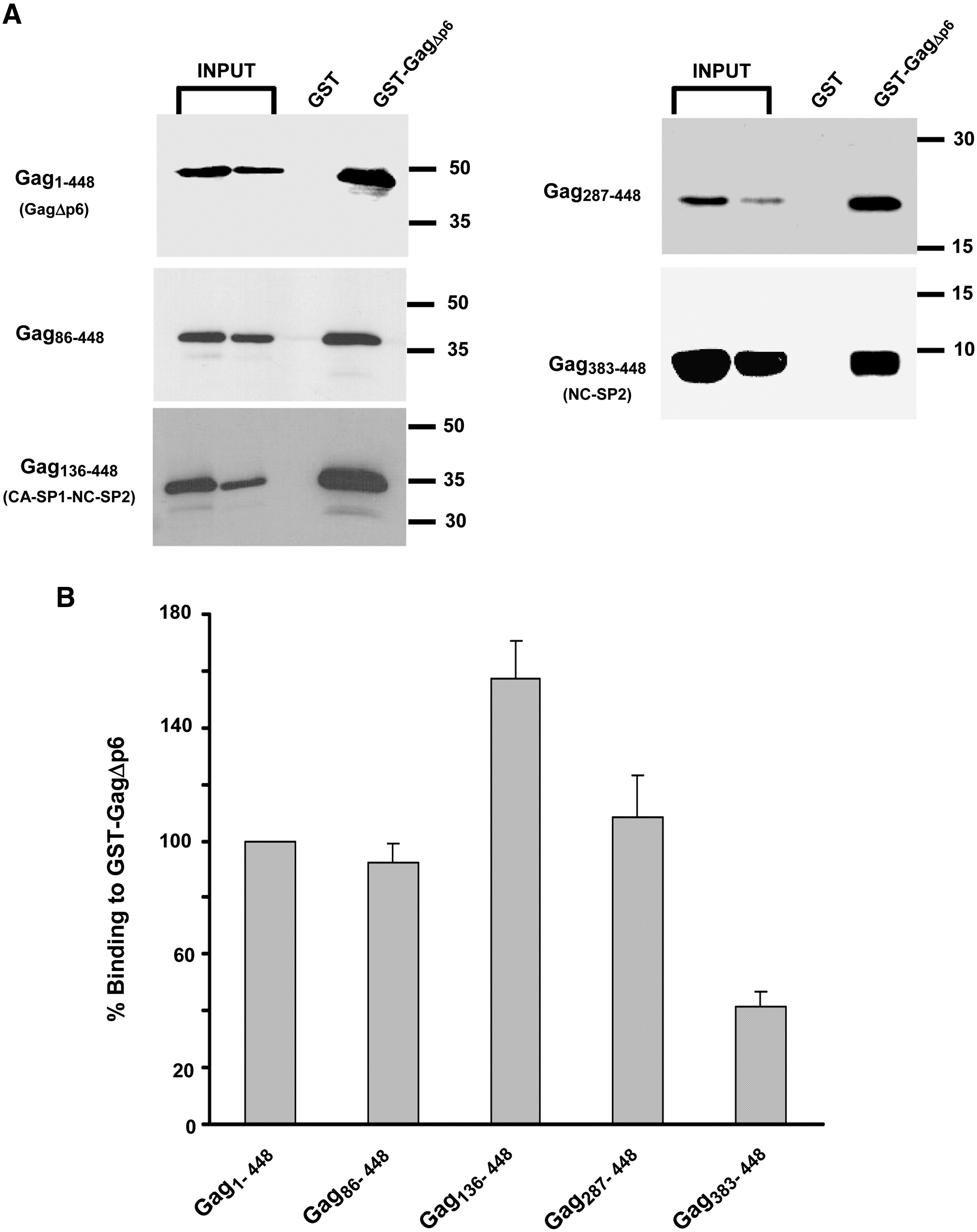
We next analyzed the ability of N-terminally truncated versions of His-SIVGagΔp6 to interact with GST-SIVGagΔp6 (Fig. 4). In contrast to the binding behavior displayed by the C-terminal truncation mutants, this set of mutant Gag proteins associated with GST-SIVGagΔp6 with efficiencies that were comparable to or even higher than that of its wild-type counterpart. The sole exception was Gag383–448 (NC-SP2 domains), whose binding capacity was 41% of that of wild-type Gag. Interestingly, mutant Gag136–448, which comprises the CA-SP1-NC-SP2 domains, exhibited a binding efficiency significantly higher than that of wild-type Gag (157.5 ± 13.2% of the wild-type value). Remarkably, the Gag mutant (Gag287–448), which only contains the C-terminal third portion of the CA and the NC, was found to interact with GST-SIVGagΔp6 as efficiently as wild-type Gag (Fig. 4B).
Binding of His-tagged SIV Gag N-terminal truncation mutants to GST-SIVGagΔp6.
Analysis of the binding capacity of SIV CA
The results described above indicated that the individual MA (Gag1–135) and NC (Gag383–448) domains were able to interact per se with immobilized GST-SIVGagΔp6, albeit with efficiencies significantly lower than that of wild-type Gag. We therefore then tested whether the mature SIV CA (Gag136–365) had the ability to associate with the Gag precursor. As shown in Fig. 5A, the CA protein did not bind to GST-SIVGagΔp6 in vitro. This result led us to speculate that the mature CA protein might exhibit more affinity for itself than for the Gag precursor. Since some retroviral CA proteins are able to form oligomers in vitro, 35 –37 we examined whether the recombinant SIV CA protein was biologically active and self-assembled in vitro. The purified His-CA protein was incubated in buffer containing 1 M NaCl for 3 h at 37°C, and the formation of CA oligomers was assessed by analyzing the pellet and soluble fractions that result from the centrifugation of the assembly reaction. Most of the SIV CA was found in the pellet fraction (Fig. 5B). Of note, an SDS-resistant protein band that, due to its electrophoretic mobility, most likely corresponds to CA dimers was also detected in the pellet fraction (Fig. 5B).

Binding of SIV CA to GST-SIVGagΔp6 and analysis of SIV CA self-assembly.
Taken together, these results demonstrate that the SIV CA multimerizes in vitro. In contrast, an SIV CA lacking the CTD (Gag136–274) was incapable of forming oligomers since this protein was only found in the soluble fraction resulting from the assembly reactions (Fig. 5C). Interestingly, analysis of the assembly reaction of the SIV CA by native gel electrophoresis revealed that this protein self-assembles into 370-kDa oligomers (Fig. 5D), which may represent the multimeric complex (CA NTD hexamers linked by CA CTD dimers) that has been described for the CA protein of HIV-1 and other retroviruses. 38
Ability of mutant Gag287–448 to interact with wild-type Gag in vivo
Since our results indicated at this point that the Gag mutant encompassing the C-terminal third of CA and the NC (Gag287–448) was the minimal Gag subdomain that interacted in vitro with GST-SIVGagΔp6 at levels similar to those of His-Gag1–448, we next asked whether this interaction would still occur in vivo. We therefore analyzed if myristoylated full-length SIV Gag (wild-type SIV Gag) could rescue mutant Gag287–448 into extracellular particles. To this end, cells infected with a recombinant vaccinia virus expressing the T7 RNA polymerase were then cotransfected with the plasmids coding for wild-type SIV Gag and for His-Gag287–448 as described in Materials and Methods.
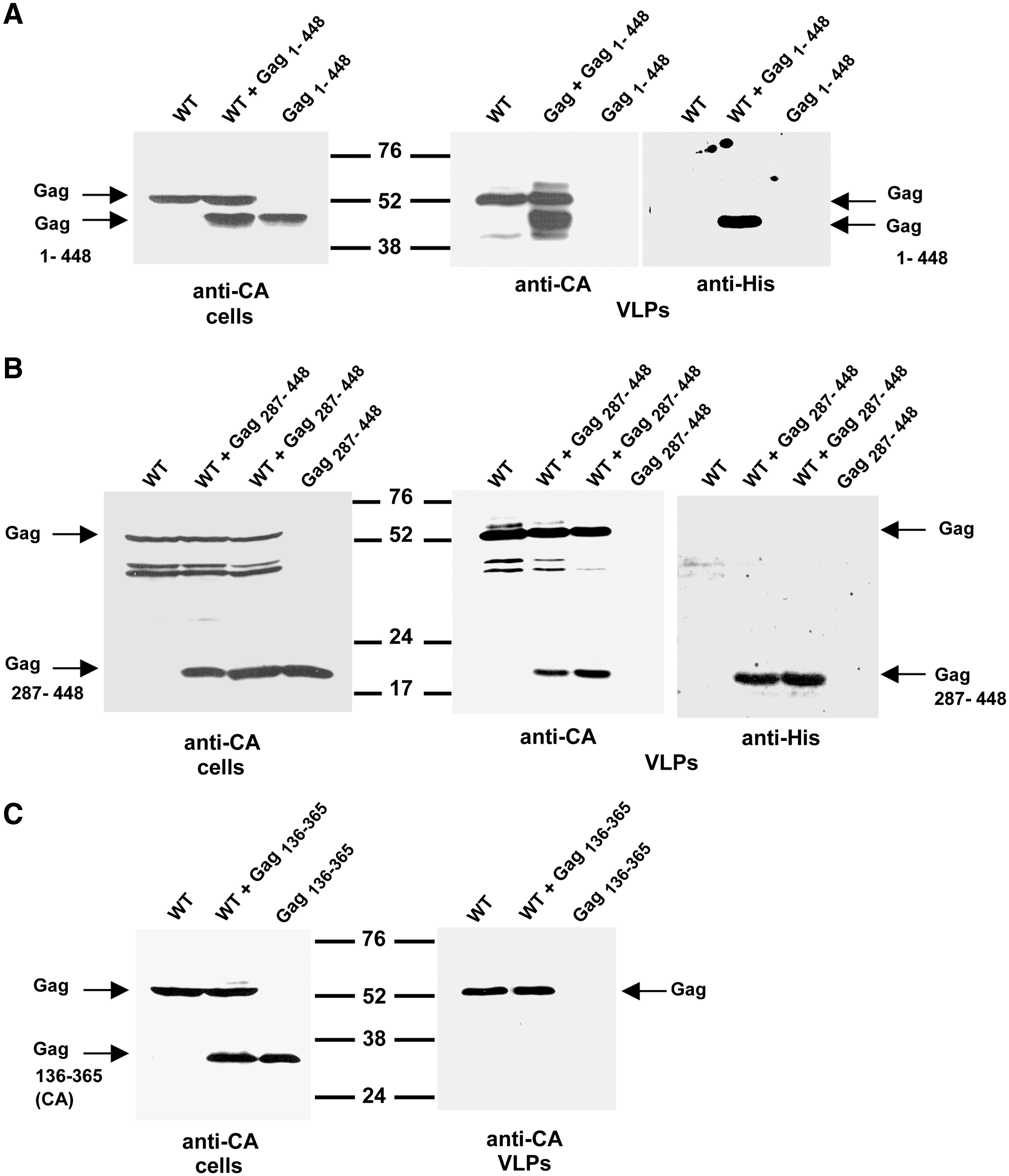
For comparison, we analyzed in parallel the incorporation of nonmyristoylated SIV GagΔp6 or SIV CA into VLPs by coexpressing wild-type Gag with His-Gag1–448 or His-Gag136–365. Cell and VLPs lysates were analyzed by Western blotting using either a monoclonal antibody to the SIV CA or the anti-His antibody. This allowed us to confirm in the cotransfection samples the identity of the His-tagged Gag-derived polypeptides. Coexpression of His-Gag1–448 with wild-type Gag resulted in the assembly of Gag VLPs containing the His-Gag1–448 protein (Fig. 6A), indicating that nonmyristoylated SIV GagΔp6 interacts in vivo with wild-type Gag.
Ability of mutant Gag287–448 to interact with wild-type Gag in vivo.
In this regard, we and others have demonstrated that a certain degree of Gag–Gag interaction and multimerization occurs prior to Gag transport to the plasma membrane. 39 –41 When we analyzed the VLPs fraction derived from cells coexpressing His-Gag287–448 and wild-type SIV Gag, we found that this Gag subdomain was also recruited into VLPs by association with wild-type Gag (Fig. 6B), which is consistent with the results obtained in the in vitro Gag–Gag interaction assays (Fig. 4B). As expected from the in vitro pull-down assays showing that the mature SIV CA was incapable of interacting with SIV GagΔp6 (Fig. 5A), we found that Gag136–365 could not be rescued into VLPs by wild-type Gag (Fig. 6C).
Quantitation in VLPs of the relative levels of wild-type Gag and His-tagged Gag proteins revealed that Gag287–448 represented up to 42% of the total amount of protein, while Gag1–448 accounted for 58% of the total protein mass in particles. This indicates that coexpression of Gag287–448 with wild-type Gag allowed this truncation mutant to make a substantial contribution to the composition of the extracellular particles.
Effect of deletions in the C-terminus of the SIV CA on particle formation
Given that the SIV Gag subdomain comprising the CA C-terminus and the NC (Gag287–448) not only proved to be highly efficient at interacting in vitro with GagΔp6, but also was recruited in vivo into VLPs by the full-length Gag precursor, we decided to further analyze the contribution of the SIV CA C-terminus to Gag multimerization. To this end, we introduced, in the context of the wild-type Gag protein, deletions within the C-terminal third of CA and SP1 (Fig. 7A), and examined the effect of these mutations on VLPs production in vivo. Gag proteins harboring deletions ΔCTD2 and ΔSP1 produced VLPs with an efficiency corresponding to approximately 30% of the wild-type value, whereas deletion ΔCTD1 caused a 5-fold reduction in particle production (Fig. 7B and C). The most drastic effect on assembly was caused by deletion of the major homology region (MHR), which reduced the ability of SIV Gag to form VLPs by approximately 90% (Fig. 7B and C).

Effect of deletions in the SIV Gag region encompassing the CA C-terminus and SP1 on VLPs formation.
Together, these data indicate that in SIV the C-terminal third of the CA and SP1 are important for efficient Gag self-interaction in vivo. Although we cannot rule out the possibility that deletion of the SIV CA MHR or adjacent sequences may affect processes other than Gag multimerization, such as Gag binding to the plasma membrane, it is most likely that a defect in Gag membrane association caused by a mutation in CA is a consequence of an impairment in protein multimerization, as has been proposed for HIV-1 Gag. 42
Discussion
Although numerous studies have contributed to a better understanding of the retrovirus assembly process, the SIV Gag sequences involved in the homotypic protein interactions that drive SIV particle formation remained to be defined. Moreover, since SIV is extensively used in vaccine development studies, the analysis of SIV biology as well as the identification of even subtle differences from HIV-1 are relevant. In the present study, we sought to map the molecular determinants in SIV Gag that mediate protein–protein interactions thereby promoting Gag multimerization. To evaluate the feasibility of developing an in vitro system to study SIV Gag–Gag interaction, we first analyzed by both pelleting assays and negative-stain electron microscopy the products formed on incubation of His-SIVGagΔp6 under fully defined conditions.
These analyses revealed that recombinant SIV GagΔp6 has the ability to self-assemble in vitro into spherical particles that are about 35 nm in diameter. Interestingly, we have recently demonstrated that recombinant full-length feline immunodeficiency virus (FIV) Gag polyprotein also assembles in vitro into particles of a similar size. 43 Notably, Campbell and Rein 34 have reported that nonmyristoylated HIV-1 GagΔp6 assembles in vitro into 25- to 30-nm spherical structures. However, the addition to the in vitro assembly reaction of cell lysates or inositol phosphate derivatives leads to the formation of HIV-1 GagΔp6 particles of a size similar to that of virions. 44
We also show here that the in vitro assembly of SIV GagΔp6 is dependent on the presence of RNA, as it has previously been demonstrated for the Gag proteins of HIV-1, 32,34 Rous sarcoma virus, 45 Mason–Pfizer monkey virus, 46 and FIV, 43 and that it is resistant to the addition of a nonionic detergent such as Triton X-100. This latter result confirms that under the experimental conditions employed in the in vitro assembly reactions, SIV GagΔp6 multimerizes into completely assembled higher-order structures, since resistance to Triton X-100 is a distinctive feature of immature particles. 39,47
To identify the regions in SIV Gag required for protein–protein interaction, we performed pull-down binding assays in which we compared the ability of a series of His-tagged Gag subdomains to bind to GST-GagΔp6 with respect to that of His-GagΔp6 considered as the wild-type construct.
By using this approach, we found that truncation of SIV Gag from its N-terminus did not adversely affect Gag–Gag interaction: indeed, Gag136–448, which corresponds to the SIV CA-SP1-NC-SP2 subdomain, exhibits a 1.6-fold higher binding activity than that of wild-type Gag, and a small Gag region consisting of the C-terminal third of the CA and the NC is able to interact with GST-SIVGagΔp6 at wild-type levels. In contrast, removal of the NC zinc-finger motifs is sufficient to reduce by 50% Gag–Gag interactions, which highlights the relevance of this SIV Gag region for protein multimerization. It is likely that in our pull-down assays the NC domain, by binding to bacterial RNA that copurifies with the recombinant proteins, facilitates Gag–Gag interactions. This is in agreement with previous in vivo studies demonstrating that the NC-RNA association promotes Gag multimerization and assembly. 23,25,31,48,49 In this regard, we found that treatment with DNase I and RNase A of both the resin-coupled GST-SIVGagΔp6 and the His-SIVGagΔp6 input reduced but did not abolish Gag–Gag interaction. Indeed, nucleases-treated Gag proteins interacted with an efficiency representing 61.7 ± 5.9% of the binding obtained with the untreated purified SIV GagΔp6 proteins (mean value ± standard deviation; three experiments).
In addition to the importance of the NC, our binding experiments also point to the crucial role that the CA domain of Gag plays in SIV Gag multimerization. The presence in the Gag truncation mutants of either the entire CA or CA-derived regions enhances Gag self-association. Indeed, the SIV Gag subdomains that correspond to CA-NC (Gag136–448) or to the C-terminal third region of CA together with the NC (Gag287–448) bind to Gag more efficiently than the NC alone. Moreover, the MA-CA protein (Gag1–365) or Gag subdomains consisting of the MA and CA regions (Gag1–231 and Gag1–306) exhibit a Gag binding capacity significantly higher than that of the mature MA.
Moreover, our in vitro binding assays allowed us to pinpoint the minimal SIV Gag subdomain capable of establishing protein–protein interactions at wild-type levels. The conclusion that the SIV Gag region spanning the C-terminal third of the CA and the NC is a major multimerization determinant is further supported by our experiments in vivo demonstrating that (1) nonmyristoylated Gag287–448 can be efficiently recruited into extracellular particles by SIV Gag and (2) internal deletions targeting the CA C-terminus and SP1 in the context of the unprocessed Gag precursor are detrimental to VLP assembly in vivo.
Although the structure of the SIV CA has yet to be determined, our results suggesting that the SIV CA C-terminus and the adjacent SP1 peptide establish crucial contacts during the formation of immature particles are in keeping with the biochemical and high-resolution structural data available for HIV-1 CA. 38,50 Interestingly, it has been proposed that the HIV-1 CA CTD dimerizes via the domain-swapping mechanism and that the swapping of the MHR segment between adjacent Gag molecules may constitute a crucial assembly intermediate. 51 This may explain the remarkable sequence conservation of the MHR in retroviruses. In this respect, our results are in line with this concept since, among the mutations we introduced into the SIV CA CTD and SP1 regions, deletion of the MHR had the most inhibitory effect on VLP production.
When we analyzed the ability of the recombinant mature SIV Gag products to interact in vitro with SIV GagΔp6, only the MA and NC proteins proved to be binding competent, with a 20% and 40% binding efficiency of that of wild-type Gag, respectively. In this regard, analogous protein-binding experiments performed with HIV-1 Gag protein did not detect an association between the MA and Gag proteins, whereas the HIV-1 NC alone displayed a binding activity similar to that of wild-type Gag. 52 In Table 1 the Gag-binding abilities of the most phenotypically relevant SIV Gag subdomains tested in our study by both GST pull-down and ELISA experiments are compared with those previously reported for HIV-1. 52
Interaction of His-tagged SIV Gag subdomains with immobilized GST-SIVGagΔp6 relative to that detected with His-SIVGagΔp6 considered as 100%. Data are from the results of the pull-down assays presented in Figs. 3, 4, and 5, whereas those in parentheses were obtained by the ELISA-based protein interaction experiments described in Materials and Methods. Values presented are averages of at least three independent experiments ± the standard deviations.
Association with GST-HIV-1Gag of Gag subdomains expressed with an N-terminal extension of the HA1 epitope of the influenza virus hemagglutinin (HA) protein. Binding activities were referred to that obtained with HA-tagged HIV-1 Gag considered as 100%. Data are from Burniston et al.52
In the case of HIV-1, these Gag constructs also included p6.
ND, not detectable.
—, not determined.
In this study, we also found that recombinant SIV CA was incapable of interacting in vitro with the Gag precursor, despite exhibiting the ability to form multimeric complexes detected by native gel electrophoresis. Notably, Zábranský et al., 53 by using the yeast two-hybrid system, could not detect an association between the HIV-1 CA and the Gag polyprotein. These results may reflect the distinct roles of the CA during virion formation: on the one hand, as a domain of the Gag precursor that participates in spherical particle assembly and, on the other hand, as a mature protein that assembles into a conical core surrounding the NC-genomic RNA complex and its associated viral enzymes. In support of this notion, biochemical and structural data obtained for HIV-1 CA indicate that there are substantial differences in the arrangements of the CA domains between immature and mature virions. 50,54
In summary, we have demonstrated that the SIV GagΔp6 is capable of assembling in vitro into spherical particles. The fact that this protein exhibited full multimerization capacity allowed us to develop an in vitro binding assay to study Gag self-interactions. The results stemming from these pull-down experiments, complemented by our studies with cells expressing SIV Gag mutants, identified the region comprising the C-terminal third portion of the CA and the entire NC as the major SIV Gag interacting domain. Our findings therefore contribute to our knowledge of the SIV Gag assembly process.
Footnotes
Acknowledgments
This work was supported by Grant PICT2005-38215 from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, Argentina) to S.A.G. M.L.R. and C.L.M. are postgraduate fellows of the National Research Council of Argentina (CONICET) and ANPCyT, respectively. J.L.A. and S.A.G. are Career Investigators of CONICET. We thank Natalio De Vincenzo (University of Buenos Aires) for assistance in the electron microscopy studies.
Author Disclosure Statement
No competing financial interests exist.