
Abstract
HIV diversity reflects multifactorial evolutionary forces, but monitoring subtype prevalence may provide clues to understanding the epidemic. In the Americas HIV-1 C is present at significant levels only in the southern states of Brazil. We describe in this study the presence of the HIV-1 C pol genome in 11.6% (95 CI 6–21%) of antiretroviral-naive individuals from São Paulo, the major city of South America, and 6.8% (95 CI 4–12%) from the second metropolitan area of the State of São Paulo, Brazil. Moreover, a significant growth trend of this subtype was documented among cases failing therapy in the area. Sequences were obtained by direct nested PCR from cDNA retrotranscribed from plasma RNA. Phylogenetic and amino acid signatures support an expansion from variants previously identified in southern Brazil. The evaluation of additional genomic regions (partial gag, envelope, and/or integrase) in samples with HIV-1 C at pol showed extensive recombination with clade B, observed in 47% of ARV-naive cases. The spread of HIV-1 C locally and to other areas of South America should be monitored as it may influence the dynamics of the epidemic.
Introduction
HIV-1 C
São Paulo, probably the core of the Brazilian AIDS epidemic, is the larger metropolitan area in South America and is located near the border of the southern states. With about 20% of the Brazilian population (
Materials and Methods
Sequence data from 1040 HIV-1-infected adult individuals referred to the Retrovirus Laboratory at Adolfo Lutz Institute for routine resistance genotype testing after failing antiretroviral therapy (ART) in the period 2005–2008 were reviewed for subtype assignment and 240 antiretroviral (ARV)-naive adult cases (newly diagnosed) were recruited specifically for this study in 2008. In the latter group, clinical data, exposure to ART, demographic information, risk transmission, and laboratory data were obtained from medical records and a specific questionnaire. Samples were shipped within 6 h to the Retrovirus Laboratory, where they were processed and cryopreserved at −70° C. HIV-1 RNA was extracted with QIAamp viral RNA Mini kit (Qiagen, Hilden, Germany). For genotypic analysis ViroSeq v2.0 (Celera Diagnostics, Alameda, CA), TrueGene (Siemens Diagnostics, USA) or previous “in house” published protocols 5 allowed the evaluation of protease (PR) and partial reverse transcriptase (RT) genes. All the genetic sequences were manually edited by proprietary software (ViroSeq or Siemens) and, for in house methods, using Sequencher 4.10 (Gene Codes, USA). HIV subtyping was evaluated at NCBI and REGA websites as a preliminary analysis.
For phylogenetic analysis, the sequences were aligned with the Clustal X program against a reference set from LANL, and the alignment was manually corrected using BioEdit software. Neighbor joining (NJ) and maximum likelihood (ML) trees were reconstructed with PAUP* 4.0b10 software according to the evolutionary model selected by ModelTest (PAUP) and viewed with TreeView. Bootstrap values were obtained after 1000 replicates. HIV-1 group O was used as the outgroup. Sequences showing outlier behavior on phylogenetic trees, discordant PR/RT classification, or a mosaic pattern at NCBI or REGA websites were analyzed by the SimPlot 2.5 program to evaluate recombination breakpoints with the following parameters: Window 300pb, Step 10, GapStrip on, Reps 1000, Kimura (two-parameter), T/t 2.0, and neighbor-joining.
Further investigations of breakpoint positions were done by inspection of subtype nucleotide signature transitions. A consensus of study sequences (60% criteria) was generated at BioEdit and was compared to Consensus B and C from NCBI. Sequences showing a clade C genome at the polymerase (pol) region were additionally sequenced at the gag, integrase, and/or envelope regions. Statistical analysis was made by the Epi Info (CDC) program using Yates corrected test or Fisher's exact test, two tailed. The respective institutional ethical committees approved the study.
Results
HIV-1 partial pol sequences from 1040 patients failing ART (2005–2008) and 230 sequences from ARV-naive patients (2008–2009) were included. Treated patients were significantly older then naive cases (41.3–35.6 years old at collection, p < 0.001). The percentage of subtype C or C mosaics (one BC and one CF) at pol among all HIV-1 infections in treated patients, by year of collection, was 0.3% in 2005 (1/329), 0% in 2006 (0/284), 1.8% in 2007 (4/220), and 2.9% in 2008 (6/205, p < 0.005). Among all naive cases, clade C at pol was observed in 8.3% of patients (19/230) (Fig. 1), which increased to 11.6% if only the São Paulo site is considered.
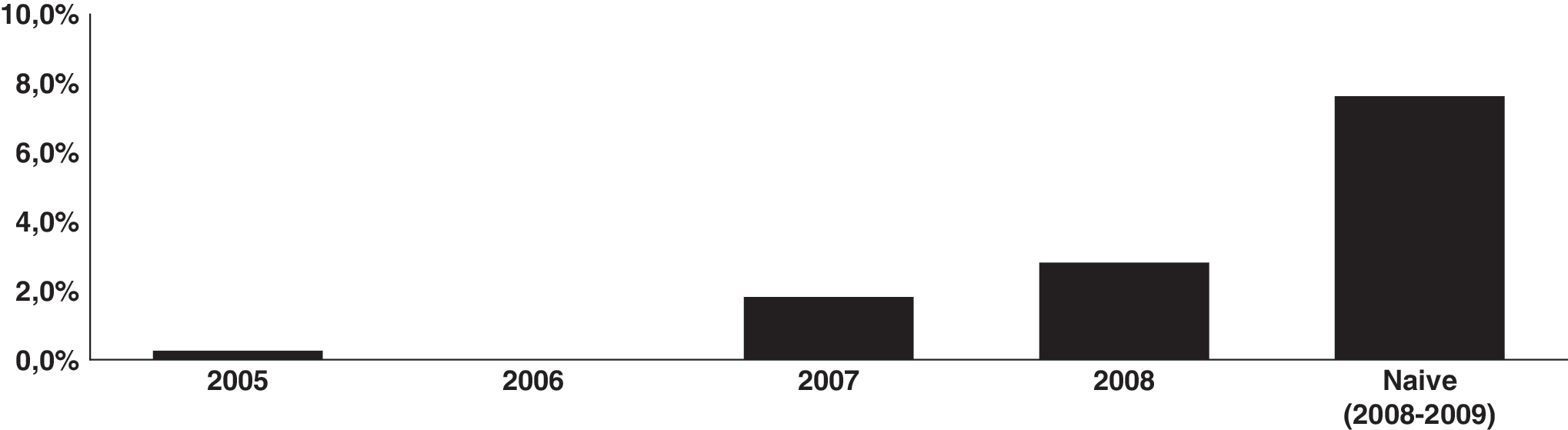
Percentage of HIV-1 pol region subtype C (or C mosaics) among patients living with HIV/AIDS (n = 1044) in São Paulo failing ARV therapy, by year of collection, analyzed from 2005 to 2008. The last column shows the proportion among ARV-naive patients (n = 230) recruited in 2008–2009 for this study.
The proportion of C or BC mosaics among naive cases is significantly higher than among treated cases overall (p < 0.0001). The significance is also present in subsets of these cases, for example, when only patients from the same metropolitan area or when only patients from 2007 to 2008 were considered (data not shown). All other sequences were subtyped as HIV-1 B, F, or BF mosaics, except for two cases, one AG_CRF02 and another clade A. All but one of the clade C pol sequences, from both naive cases and patients failing ART, grouped within a southern Brazil subtype C cluster at the phylogenetic tree (Fig. 2). The only HIV-1 C pol sequence from treated cases not clustering within the Brazilian group (sample 57_08) is from a male patient from Luanda, Angola.

Representative neighbor-joining phylogenetic tree of partial pol region sequences of the study (in bold), generated with the GTR + I + G model, using Paup 4.0. Reference HIV-1 subtypes were obtained from the Los Alamos HIV Sequence Database including HIV-1 C sequences from southern Brazil. Bootstrap values at key branches are depicted. Brazilian sequences from the study are designated (in bold) by BR, Brazil; year of collection; city of origin (CAMP, Campinas; SP, São Paulo; Southern POA, Porto Alegre; ITJ, Itajaí; and CTB, Curitiba) and ID number (e.g., BR06SP20).
Information on risk factors was obtained from naive cases, where 25% had one or more partners using ARV therapy; in five cases women also used MTCT prophylaxis. Among 168 volunteers without a known partner on ARV therapy, about half were men who have sex with men (MSM). MSM from São Paulo were more likely to be infected with clade C then MSM from Campinas (16% vs. 3%, p < 0.04) and, in this latter site, the proportion of clade C among heterosexuals, whether male or female, was 14% compared to 3% among MSM (p < 0.03). We did not found a correlation between HIV clade C and gender or age. Anal sex was more frequent among cases infected with non-C clades (64% vs. 50%), but the difference was not significant (p = 0.4). When we study the phylogenetic relationship of São Paulo HIV-1 clade C sequences with those from southern Brazil with known risk factors (heterosexual or MSM) we do not find risk-related clusters by phylogenetic analysis.
Out of the 19 naive cases with the clade C genome at pol, 17 had one or more additional regions (gag, integrase, and/or envelope) sequenced. Mosaics were common, and 47% (8/17) showed different CB mosaic structures (Fig. 3). One sequence (366_09) had breakpoints at pol similar to that of CRF31_BC, clustering with these CRF references (Fig. 3) but showing an additional non-C segment at gag not present in the CRF31_BC. Amino acid signatures at the pol genomic region were analyzed. Comparing the HIV-1 C consensus (international, southern Brazil, and São Paulo), some amino acid signatures were common both in southern Brazil and in São Paulo sequences that distinguished them from the international consensus (S12T, I19L, N37K, K41N at PR; E39D and I135T/M at RT). In this region, the substitution A173T is present in the southern Brazil consensus but, albeit present in some samples, is not present in the São Paulo consensus. At the G123 residue the southern Brazil consensus has a D whereas in São Paulo it has an N (Fig. 4).
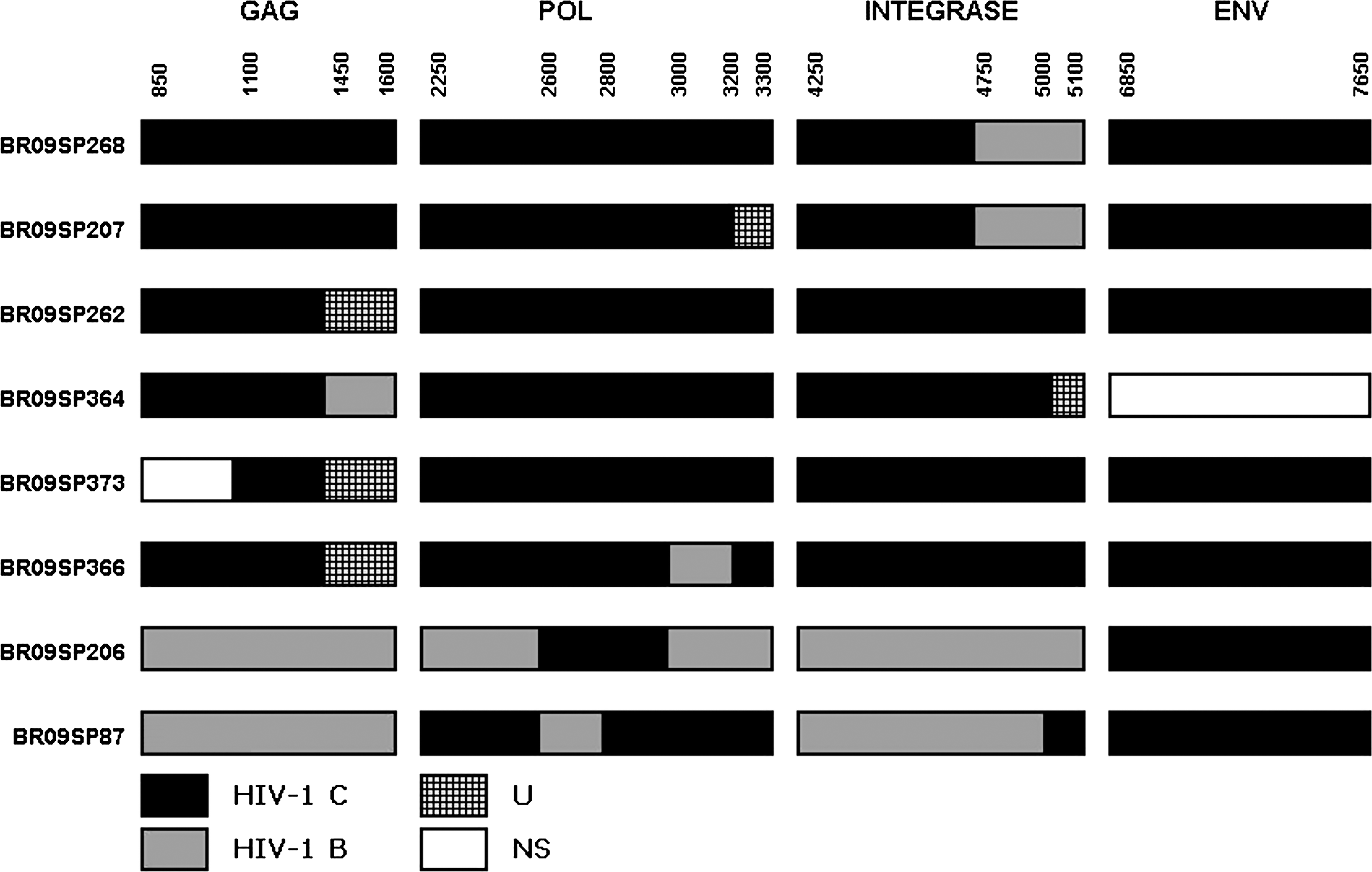
HIV-1 sequence breakpoint positions (according to HXB2 reference) of mosaics at the partial gag, pol, integrase, and envelope regions. The approximate breakpoint position was deduced from SimPlot analysis. U, unknown, subtype not defined; NS, not sequenced.

HIV-1 amino acid alignment from protease (top) and partial reverse transcriptase (bottom) of Consensus sequences for subtype B and C international (from NCBI), Consensus (60%) from southern Brazil, and study HIV-1 C sequences.
Sequences are available at GenBank (accession numbers: HM024737–HM025942, HM627515–HM627517, HM533970–HM534205, HM854664–HM854708, EU189705–EU189708, EU189710–EU189716, EU189700, EU189702, EU189717–EU189720, EU189722, EU189724–EU189727, and EU189729–EU189734).
Discussion
São Paulo, the sixth largest metropolitan area in the world, is probably a major core for the mostly HIV-1 B Brazilian AIDS epidemic. In this study, we document for the first time evidence of a significant increase in the proportion of the HIV-1 C subtype among HIV-infected adult individuals in São Paulo city, with over 10% of clade C among ARV-naive cases. We also observed a similar important presence of HIV-1 C among ARV-naive cases in the Campinas metropolitan area, the second largest city of São Paulo state, 80 km from São Paulo city.
To better contextualize these findings, we evaluated retrospectively all cases of ARV-exposed adult patients failing ART genotyped for resistance mutations in our laboratory from 2005 to 2008. A significant trend of an increasing proportion of HIV-1 C was observed, supporting the findings among naive cases. The proportion of HIV-1 C genomes was significantly higher when MSM from São Paulo is compared to those in Campinas city. In the latter, clade C predominated among heterosexual cases. This may suggest distinct dynamics of the epidemic, but the sample size limits this evaluation.
We also tried to identify phylogenetic relationships among two major risk groups, MSM and heterosexuals, comparing sequences from São Paulo state with cities in southern Brazil. Our sequences did not show clear risk-related clusters, and accurate risk information was limited. Moreover, the frequency of bisexual behavior among MSM in Brazil would itself make it difficult to find clear clusters.
Overall, as can be seen in Fig. 2, sequences from southern Brazil and from São Paulo form a subcluster as previously shown for Brazilian C sequences. The origin of the infections by HIV-1 C is unclear, but because the city of São Paulo hosts many sociocultural events, has a large population, and has an economic position that would naturally represent the major entry point for a new variant in the area; however, only phylodynamic studies can evaluate this more precisely.
We sequenced additional segments of most HIV-1 C pol isolates from naive cases and, albeit sequencing only part of the genome, about half showed evidence of intersubtype recombination. The high proportion of BC mosaics, representing 47% of cases with more than one region sequenced, suggests that the B and C epidemics have been intermixed for some time in the area.
HIV-1 C is the most prevalent HIV-1 variant worldwide, and much work has been dedicated to document potential factors associated with its transmission or differential pathogenesis. A fitness advantage in acquisition has been proposed since the late 1990s, and more recently 8 it was observed that subtype C isolates, as compared to subtype A, display a higher transmission efficiency using ex vivo cervical tissue derived from organ culture. Most studies, however, do not support a differential transmissibility, 9 which suggested that the variant may have less ability to replicate in peripheral blood mononuclear cells (PBMCs) but no distinct transmissibility. Evaluating sexual risk factors associated with transmission of clade C or B in a small study in Porto Alegre, southern Brazil, where clades B, C, and CRF31_BC circulate in similar proportions, it was observed that infection with clade B as compared to clade C was more dependent on anal sex. 10 In the present study the proportion was more comparable and the difference was not significant. It is conceivable that the variant is only there, at the right place, taking advantage of a favored socioepidemiological context, but this issue is far from settled.
The presence of HIV-1 C in Brazil has been an issue since the identification of clade C in samples from Porto Alegre in the early 1990s during the WHO isolation and characterization global initiative. 11 In a study in 2002, Martinez et al. 4 described an important presence of the variant in a city in south Brazil, near the border with Uruguay. This and other neighboring countries, such as Argentina, along with most Brazilian cities, had shown only sporadic identification of clade C. 2,7
In central Brazil, clade C has been reported in a few cases, below 4%. 3 In Rio de Janeiro a study at a local blood bank identified one sample (1.4%) of a BC mosaic. 12 In São Paulo, a study from our group identified a low proportion (0.5%) in samples from 1998–2003, which represented 2.3% if only ARV-naive cases were considered. 13 In another study from a blood bank in 2006, 3.8% of clade C was observed 6 and in a study in 2007 with naive patients enrolled from this blood bank, clade C was identified in 5.7% of cases. 14
More recently, a study with ARV-naive cases in six cities in Brazil documented HIV-1 C in 6% of cases from São Paulo, a slightly larger proportion then that observed in other sites outside south Brazil. 15 In the nearby city of Santos, a recent study based on 33 cases documented a prevalence of 6% (2/33), with figures of 1.8% and 2.1% in evaluations for the years of 2005–2006. 16 Consistent with these findings, our study documents a trend for an increase in the proportion among chronic infected cases failing ARV therapy and a relatively high proportion of clade C among naive cases in the major metropolitan areas of São Paulo.
The molecular information available suggests a phylogenetic relationship with amino acid signatures common to other southern Brazil HIV-1 C isolates. The only exception was traced to acquisition of HIV-1 abroad. This highlights the need for proper contextualization of data sets when the dynamics of the epidemic are studied using available sequences, as some may lack adequate epidemiological information. The spread of HIV-1 C to other areas of the country is expected and its surveillance should be implemented to both monitor any potential impact of this cocirculation in the national epidemic as well as to better understand the role of viral diversity in the pathogenesis of HIV.
Footnotes
Acknowledgments
This work was supported by FAPESP, Grant 2006/61311-0. We thank the São Paulo Secretary of Health, through the São Paulo Aids Program, for the collaboration. We also thank R.B. Andrade, A.F.A.C. Siqueira, F.P. Carvalho, J.G. Batista, and A.J. Silva for their contribution. We are grateful to the volunteers and the staff at the STI clinics involved in the study. The São Paulo HIV Salvage Workgroup collaborators include R.A.M.B. Almeida, C.M.P. Vazquez, D.M. Ferreira, Jr., L. Jamal, I.O. Silva, P.E. Braga, L.C. Pereira, Jr., and L. Hornke.
Author Disclosure Statement
No competing financial interests exist.