
Abstract
We investigated the current molecular epidemiological status of HIV-1 in Mongolia, a country with very low incidence of HIV-1 though with rapid expansion in recent years. HIV-1 pol (1065 nt) and env (447 nt) genes were sequenced to construct phylogenetic trees. The evolutionary rates, molecular clock phylogenies, and other evolutionary parameters were estimated from heterochronous genomic sequences of HIV-1 subtype B by the Bayesian Markov chain Monte Carlo method. We obtained 41 sera from 56 reported HIV-1-positive cases as of May 2009. The main route of infection was men who have sex with men (MSM). Dominant subtypes were subtype B in 32 cases (78%) followed by subtype CRF02_AG (9.8%). The phylogenetic analysis of the pol gene identified two clusters in subtype B sequences. Cluster 1 consisted of 21 cases including MSM and other routes of infection, and cluster 2 consisted of eight MSM cases. The tree analyses demonstrated very short branch lengths in cluster 1, suggesting a surprisingly active expansion of HIV-1 transmission during a short period with the same ancestor virus. Evolutionary analysis indicated that the outbreak started around the early 2000s. This study identified a current hot spot of HIV-1 transmission and potential seed of the epidemic in Mongolia. Comprehensive preventive measures targeting this group are urgently needed.
Introduction
M
The Second Generation HIV/Sexually Transmitted Infections (STI) surveillance program (SGS) for HIV/STI serological studies in various behaviors was initiated in Mongolia in 2002. According to the latest results of the SGS conducted in 2007, the HIV prevalence among blood donors and pregnant women was zero and among 1350 tuberculosis patients was 0.15% (95% CI 0.00–0.35). 1 The results of our 2007 prevalence survey of 2465 individuals (1415 high-risk and 1050 healthy control populations) in Mongolia demonstrated that the current HIV prevalence is low, but according to the high prevalence of syphilis (anti-TP 23.1%) and HCV (anti-HCV 8%) in high-risk populations, the risk status for HIV-1 infection is estimated to be high. 2 The high-risk populations included FSW, MSM, mobile men, tuberculosis (TB) patients, and male STI clinic clients; healthy control populations were youth and blood donors.
Knowledge about current patterns and trends of HIV infections is essential for planning and evaluating prevention programs and for resource allocation. In the past, epidemiological data on newly diagnosed HIV/AIDS and results of a surveillance study of HIV/STI have been used for planning and targeting HIV prevention programs in Mongolia. However, these data were not sufficient to implement a comprehensive, effective, targeted strategy for prevention of HIV infection in Mongolia. To gain a better understanding of the current HIV status, a more detailed molecular epidemiological study using phylogenetic analyses is needed. Molecular epidemiological analyses are useful tools to gain information about the origin of HIV epidemics and transmission patterns. 3,4 Moreover, information regarding the genetic diversity of HIV strains, and the geographic prevalence of genotypes, is important for the evaluation of diagnostic tests and vaccines and for their effective application. 5,6
In the present study, we used phylogenetic analyses to determine the HIV-1 subtypes circulating in Mongolia, especially the dominant subtypes responsible for the outbreak in the infected population. Then, we performed a Bayesian coalescent-based framework to investigate the origin and estimate the onset year of the dominant HIV-1 subtypes in Mongolia.
Materials and Methods
Study population
After obtaining informed consent, blood samples were collected anonymously from 39 Mongolian HIV-1-positive patients attending the National Center for Communicable Diseases of Mongolia (NCCD) in November 2007 and May 2009. For all of them, HIV infection was diagnosed from 1997 to 2009, and all participants were infected through sexual contact. In this study, we also examined two stored serum samples at the NCCD obtained from a Russian married couple who were diagnosed with HIV-1 in Mongolia in 2007. Therefore, a total of 39 (69.6%) samples out of all 56 reported cases in Mongolia as of May 2009 was examined in this study. The study subjects were 32 men [mean age (±SD): 31.3±7.8 years, range: 17–52 years] and 9 females (25.9±5.1, 18–33 years) and 63.4% of them had a high level (college/university) of education (Table 1). The StatView software version 5.0 (SAS Institute, Cary, NC) was used for statistical analyses. The collected serum samples were sent to the AIDS Clinical Center, National Center for Global Health and Medicine (NCGM), Tokyo, Japan, for further analysis. The study protocol was approved by the ethics committees of NCGM (H19-448 and H20-545) and the Ministry of Health, Mongolia (2007-#7 and 2008-#3).
Five (55.6%) of the female cases were female sex workers.
Amplification and sequencing of HIV-1
Total RNA was extracted from 140 μl of serum, using the QIAamp RNA Mini Kit (Qiagen, Valencia, CA) according to the instructions supplied by the manufacturer. HIV-1 cDNA was obtained by reverse transcriptase polymerase chain reaction (RT-PCR) using the TaKaRa One Step RNA PCR kit (AMV, TaKaRa Bio Inc., Japan) and then DNA fragments were amplified using the TaKaRa Ex Taq Hot Start Version (TaKaRa Bio Inc, Japan) with the following primer sets. A total of 1065 bp of the polymerase (pol) fragment (HXB2: 2243–3308) containing the regions encoding the full protease and first 560 nucleotides of RT was amplified by RT-PCR with primers of F-1849 (5′-GAT GAC AGC ATG TCA GGG AG-3′) and R-3500 (5′-CTA TTA AGT CTT TTG ATG GGT CAT AA-3′). DRPRO5 (5′-AGA CAG GYT AAT TTT TTA GGG A-3′), DRPRO2L (5′-TAT GGA TTT TCA GGC CCA ATT TTT GA-3′), DRRT1L (5′-ATG ATA GGG GGA ATT GGA GGT TT-3′), and DRRT4L (5′-TAC TTC TGT TAG TGC TTT GGT TCC-3′) primer sets were used for nested PCR. A total of 447 bp of the envelope (env) fragment (HXB2: 6834–7281) containing the region C2V3 was amplified by RT-PCR with primers of V1V2-1 (5′-TGT GTA CCC ACA GAC CCC AAC CC-3′) and IC462M (5′-GCC CAT AGT GCT TCC TGC TGC T-3′). ENV02 (5′-ATG GTA GAA CAG ATG CAT GA-3′) and E115 (5′-AGA AAA ATT CCC CTC CAC AAT TAA-3′) primer sets were used for nested PCR. The amplified DNA was purified using the QIAquick PCR purification kit (Qiagen Inc.) according to the protocol provided by the manufacturer. Purified DNA was sequenced by using the ABI BigDye Terminator v3.1 cycle sequencing ready reaction kit (Applied Biosystems, Foster City, CA) and processed with an automated ABI 3730 DNA Analyzer (Applied Biosystems).
HIV-1 subtype determination and distance-based phylogenetic inference
Analyses of HIV-1 subtype and circulating recombinant form were performed using the REGA HIV-1 Subtyping Tool.
7
The basic local alignment search tool (BLAST) (
Bayesian coalescent inference using the subtype B sequences
Evolutionary rates, molecular clock phylogenies, and other evolutionary parameters were estimated from heterochronous genomic sequences of subtype B in Mongolian, non-Mongolian, and some reference sequences by using the Bayesian Markov chain Monte Carlo (MCMC) method. The reference sequences were obtained from the HIV sequence database in the Los Alamos National Laboratory with their sampling time. The reference alignment set for the pol region consists of 22 sequences of subtype B viruses covering Korea, China, Japan, Russia, Europe, and North America, with the sequence of D.KE.97.ML415_2 as the subtype's outgroup. The set for the env region consists of 20 sequences of subtype B viruses covering Korea, China, Japan, Russia, Europe, and North America, with the sequence of C.US.98US_MSC5016 as the subtype's outgroup. Each reference set was piled up with the Mongolian sequence analyzed in the present study and realigned using CLUSTAL-W. The following analyses were performed in each region of the sequence alignment. The nucleotide substitution model used in the analyses was evaluated by the hierarchical likelihood ratio test using PAUP v4.0 beta 9 with MrModel test, 10 and the general time-reversible (GTR) model 11 with both invariant sites (I) and gamma-distributed site heterogeneity (G) with four rate categories had maximum likelihood. Bayesian MCMC analyses were performed by BEAST v1.4.8 12 using the GTR+I+G and a relaxed molecular clock model (the uncorrelated lognormal-distributed model). 13 Three different population dynamic models, Exponential growth, Logistic growth, and Bayesian Skyline Plot (BSP), were tested in the analyses, and the exponential model was adopted as the most likely phylogeny according to the BSP property. Each Bayesian MCMC analysis was run for 30 million states and sampled every 10,000 states. Posterior probabilities were calculated with a burn-in of 4 million states and checked for convergence using Tracer v1.4. The maximum clade credibility tree for the analyzed set of the MCMC data was annotated by TreeAnotator in the BEAST package. The posterior distribution of the substitution rate obtained from the heterochronous sequences was subsequently incorporated as a prior distribution for evolutionary rate of the pol region as well as the env region, thereby adding a timescale to the phylogenetic histories of the given viruses and enabling the estimation of the times of the most recent common ancestors (tMRCA). 14
Results
The pol region (1065 bp) was successfully amplified and sequenced in 41 sera. According to the REGA HIV-1 Subtyping Tool, the distribution of HIV-1 genotypes in the study population was as follows: 32 cases (78%) of subtype B, two (4.9%) subtype C, two (4.9%) subtype G, four (9.8%) CRF02_AG, and one case (2.4%) of CRF01_AE.
To investigate the geographic origin of the Mongolian strains, the 41 pol sequences were compared against all HIV-1 sequences in the NCBI database using a BLAST similarity search. A distance-based phylogenetic tree (NJ tree) was constructed with 67 pol reference sequences (Fig. 1A). As shown in the phylogenetic tree of the pol gene region, the majority (78%) of the sequences belonged to subtype B, in which two distinct clusters, named cluster 1 and cluster 2, were identified. Twenty-one (65.6%) of the total 32 subtype B sequences, which included 16 MSM, two HSM, and one HSF Mongolians, were grouped into “cluster 1.” This cluster showed a remarkable monophyly with a long branch against the other sequences and high bootstrap value (<98%). The mean nucleotide diversity within the cluster was low (<0.01), indicating that members of this cluster were closely related. Russian B strains (07RUS-K0511NOV07 and 07RUS-K0611NOV07) were included in cluster 1, suggesting a Russian carrier was responsible for the Mongolian HIV-1 epidemic. Cluster 2 consisted of sequences from eight (25%) MSM Mongolian patients and four reference strains from Korean subtype B origin. Cluster 2 was more divergent than cluster 1, and had a relatively low bootstrap value (<90%). We also identified three small groups, group 2a, 2b, and 2c, in cluster 2. They had high clade credibility (bootstrap value >98%) and low genetic divergences (<0.01).
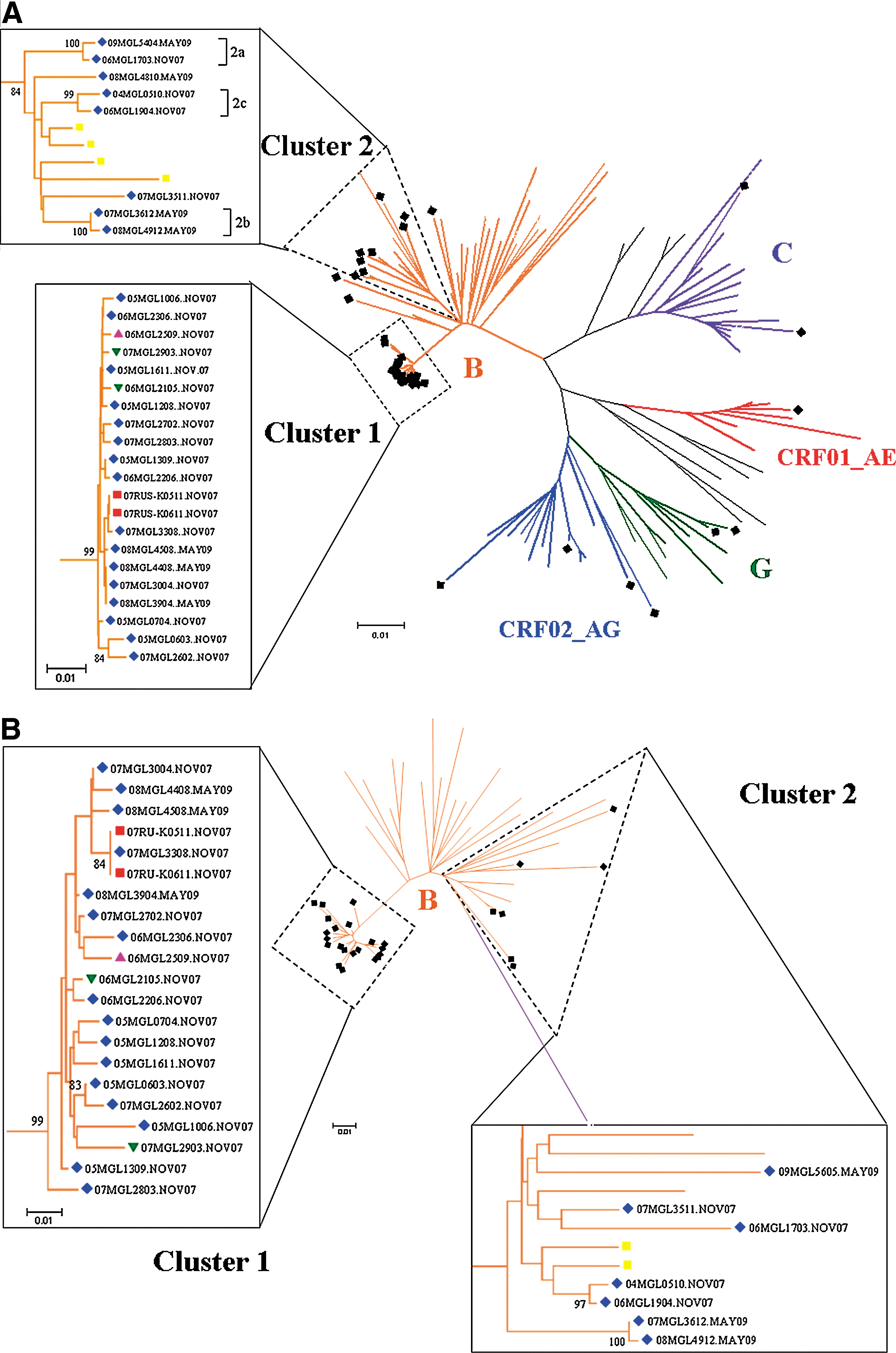
Distance-based phylogenetic tree. Unrooted radial phylogeny of
Two sequences isolated from two Mongolian heterosexual men belonged to subtype G. They were close relatives and seemed to have diverged from the reference sequences of subtype G of Central African (Nigeria) origin. All other remaining Mongolian sequences were from female patients. Two sequences (one was FSW) were close to the reference sequences subtype C of Asian (India) and Northeast African (Ethiopia and Burundi) origins, respectively. The sequences of four sex workers represented CRF02_AG. These sequences were close to reference strains isolated from Uzbekistan, Cameroon, and Ghana. The remaining isolated sequence was CRF01_AE, which was close to reference strains from Vietnam.
Twenty-eight out of the 32 subtype B samples based on the pol gene sequences were successfully amplified and the env (C2V3) gene region was sequenced and a phylogenetic tree was constructed (Fig. 1B). All samples were also classified as subtype B, indicating that they were not intersubtype recombinant forms if judged from the pol and env genes. Since the env gene region of HIV-1 has high sequence diversity, it is suitable for gene evolution analysis. Nevertheless, the intragroup nucleotide diversity of all 21 sequences on the env gene region in cluster 1 was very low, reflecting the rapid expansion of transmission of this lineage of HIV-1 in Mongolia. In contrast, seven other sequences on the env gene region belonging to cluster 2 were considerably divergent, suggesting a multiple origin of cluster 2.
Newly diagnosed cases of HIV-1 infection in Mongolia markedly increased in 2005 (Fig. 2, upper panel). Since that year, 7–13 new cases of HIV-1 infection had been diagnosed annually. Although the dominant subtype in Mongolia is currently subtype B, the first subtype-confirmed case in our analysis was CRF02_AG and was detected in 1997 (Fig. 2, lower panel). In 2004, one virus was classified into subtype B. This virus belonged to cluster 2, and was assumed to be of Korean origin. In 2005, viruses from six patients were classified as subtype B while one was subtype C and CRF02_AG, respectively. All subtype B viruses belonged to cluster 1 in our phylogenetic analysis. In 2006, viruses from six patients were also classified as subtype B, and two of them belonged to cluster 2. One of these cluster 2 viruses was also assumed to be of Korean origin (i.e., group 2c). From 2006, cluster 1 viruses had been twice as dominant as cluster 2 viruses. Other subtypes and CRFs were rarely collected in Mongolia.
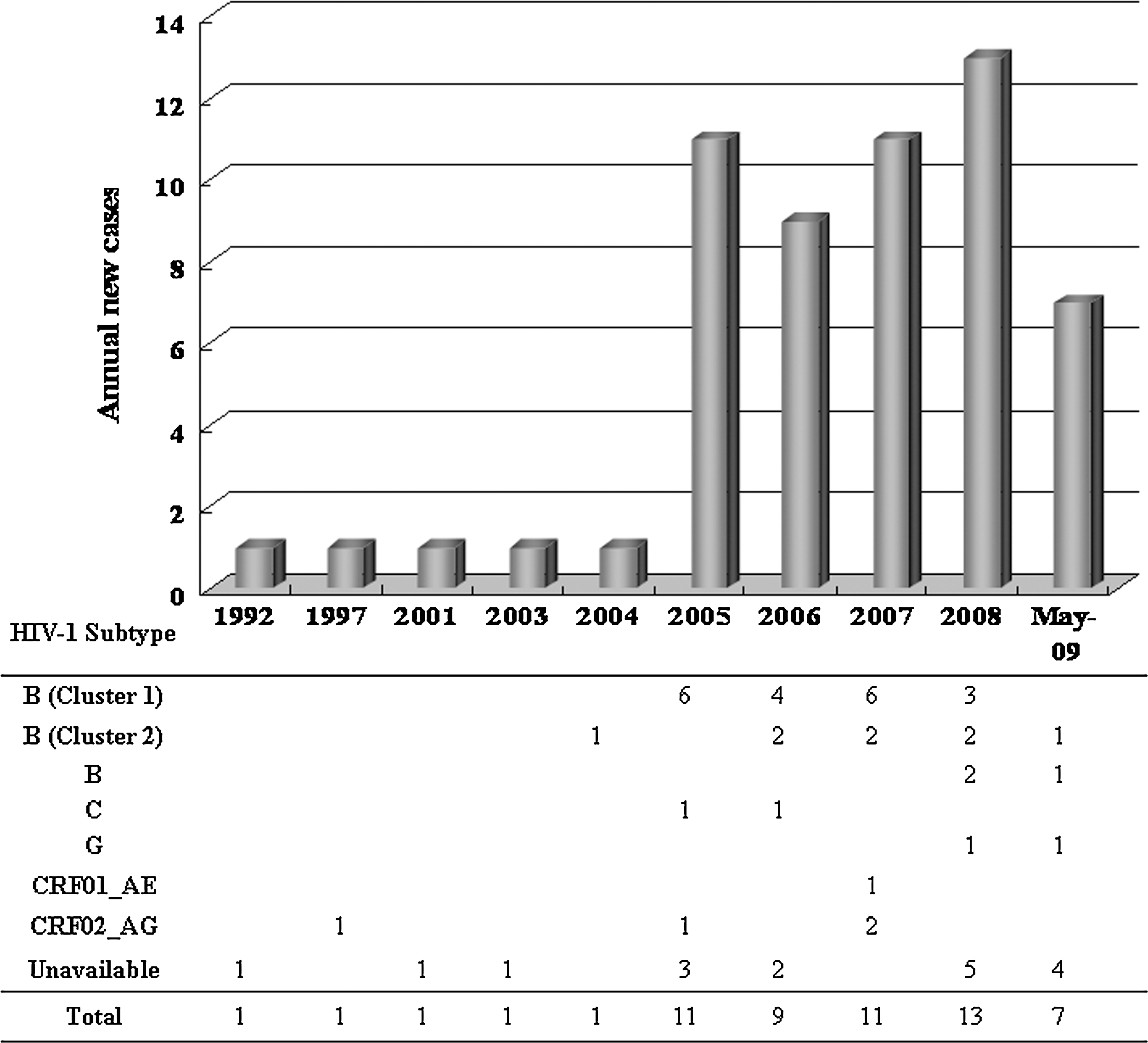
Year-diagnosis rates of HIV-1 infection in Mongolia between 1992 and May 2009 (upper panel). The number of patients and their identified HIV-1 subtypes are listed in the lower panel.
To assess the result of the distance-based analysis, and to estimate tMRCAs of the Mongolian clusters, we performed a Bayesian coalescent-based phylogenetic inference. Appling the Bayesian relaxed molecular clock method to the phylogeny, the estimated mean evolutionary rates per year per site were 1.90×10−3 and 6.66×10−3 for pol and env, respectively (Supplementary Table S1; Supplementary Data are available online at
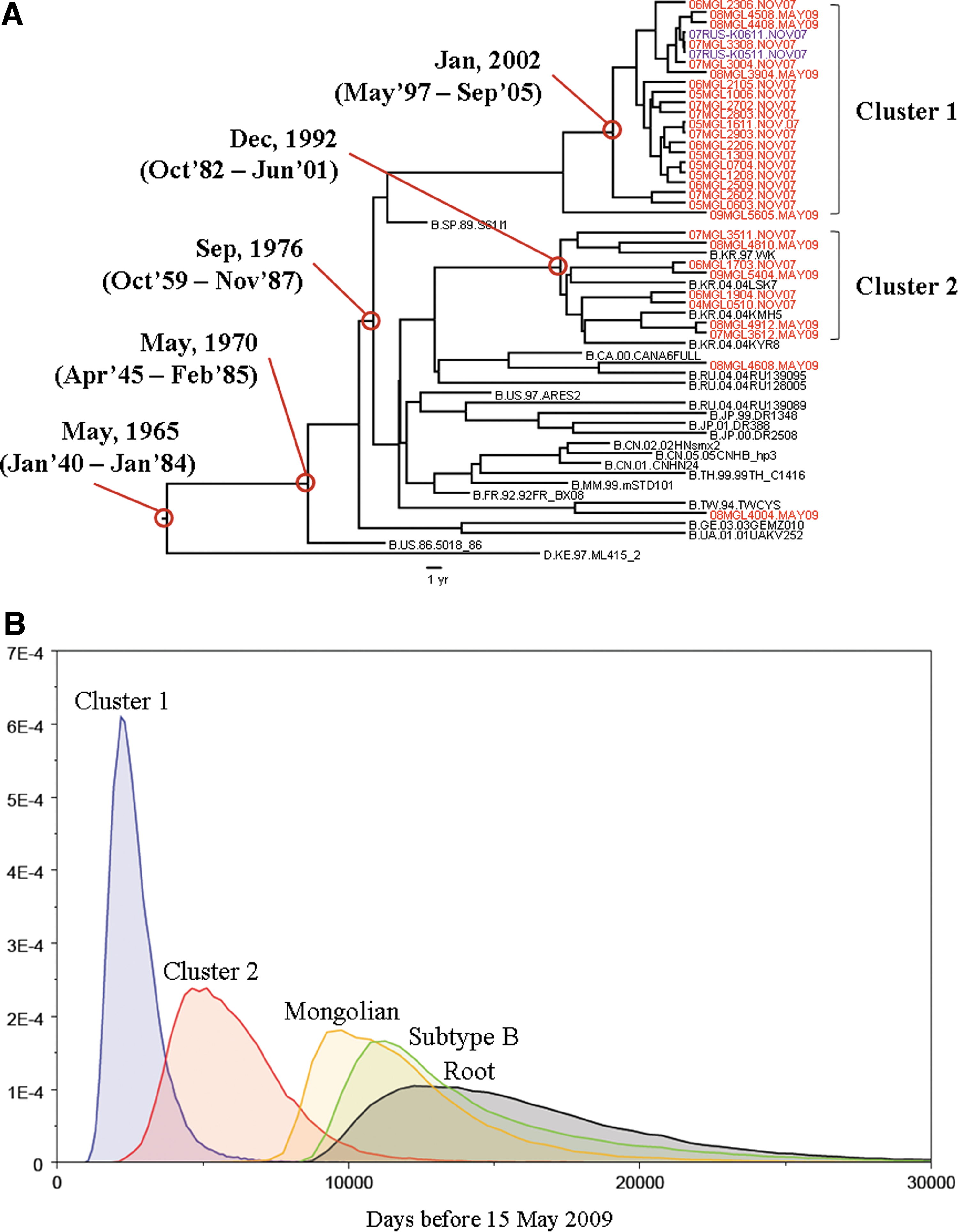
Bayesian coalescence analysis of pol gene HIV-1 subtype B.
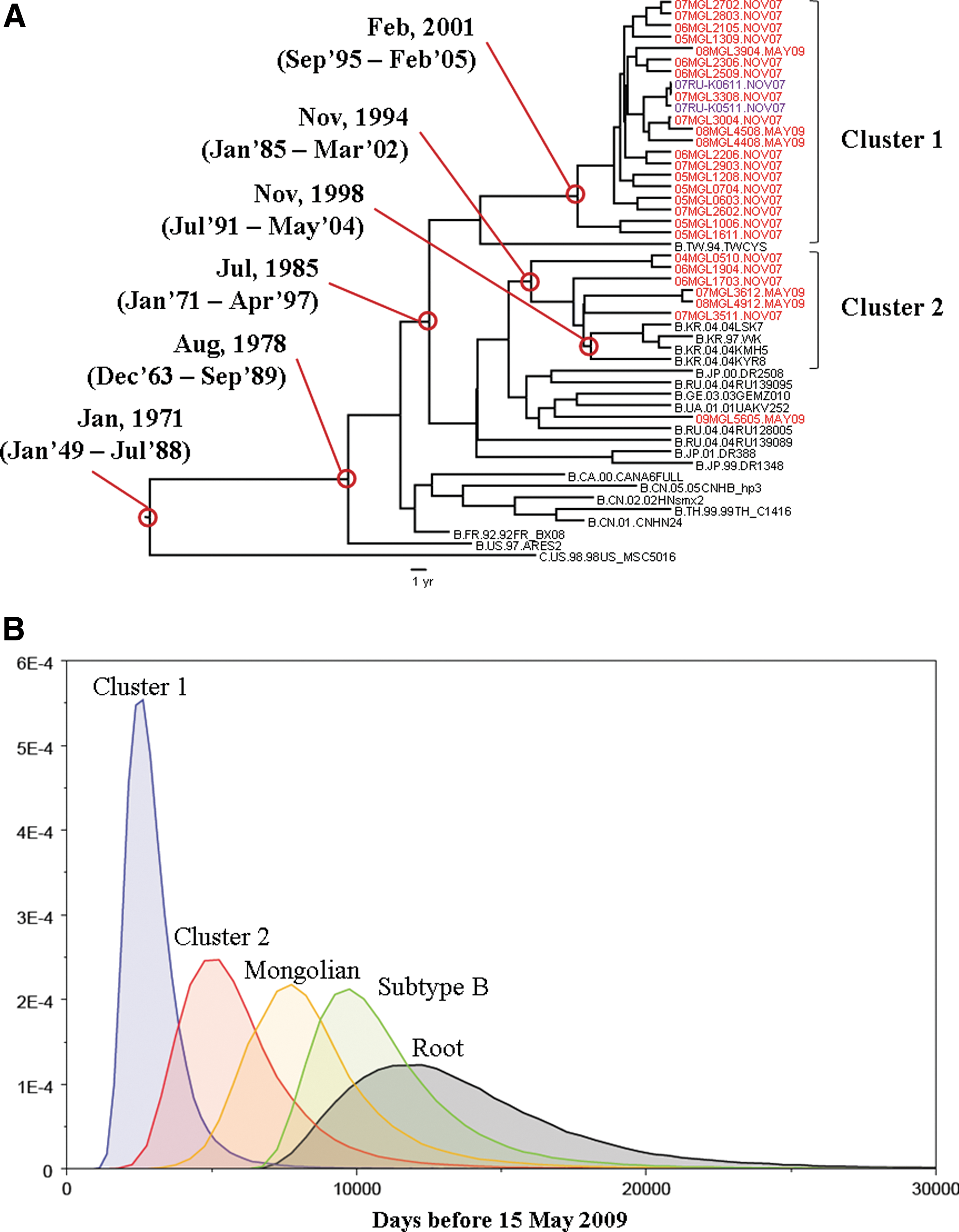
Bayesian coalescence analysis of the env gene of subtype B.
Discussion
It is important to monitor circulating subtypes and the emerging genetic diversity of HIV-1 not only because it has implications for global surveillance but also because it should facilitate risk analysis of HIV-1 transmission and help effective strategies for HIV-1 prevention. 15 –19
The present study is the first report that provides definitive evidence of HIV-1 infection occurring in a low prevalence country, Mongolia. Our results showed that HIV-1 subtype B is responsible for nearly 78% of the analyzed samples, and possibly by sexual network within the predominant MSM (84.8%) risk group. The phylogenetic analyses of HIV-1 pol subtype B sequences from Mongolian and non-Mongolian origins showed that sequences of cluster 1 and cluster 2 formed monophyletic groups compared with other viruses of the same and different subtypes from around the world, indicating that HIV-1 subtype B entered Mongolia through two distinct origins.
The most intriguing feature of this epidemic is the very low genetic diversity of cluster 1. Molecular analysis strongly indicated that HIV-1 spread rapidly during a relatively short period with the same ancestor virus. Patients of cluster 1 were diagnosed between 2005 and 2008. However, the result of the Bayesian MCMC analyses suggests that the main outbreak occurred around the early 2000s. The short-term expansion also strongly suggests a high-risk sexual behavior in this population. 20 Although most patients were MSM, the group also included bisexual and female patients. They could potentially serve as a bridge between MSM and a lower-risk population, such as heterosexually active adults. Based on the extremely high prevalence of syphilis in FSW as determined in our nationwide surveillance of HIV/STI in 2007, 2 Mongolia is at high risk of an expansion of HIV-1 infection. In Russia, subtype B is not a major subtype in the total HIV epidemic but is predominant among MSM. 21 However, it is possible that the Russian subtype B strain may become the major strain in the Mongolian population in the future. Based on this, comprehensive preventive measures are urgently needed for this group and our team has already started taking action.
The median evolution rates estimated for Mongolian subtype B in the pol (1.9×10−3 substitution site per year) and env region (6.66×10−3 substitution site per year) were comparable with the rates reported previously for these genomic regions for subtype B in other countries (pol 2.5×10−3 substitution site per year, 22 env 5–7×10−3 substitution site per year 23,24 ). Considering these evolutionary rates, the older origin was probably from Korean HIV-1 subtype B, and first emerged in Mongolia around the early 1990s, almost a decade before the first detection of HIV-1 subtype B in Mongolia. However, this group also has the potential to be a major cluster in the future. This conclusion is based on the demographic data that documented that most of the patients in this group lived in South Korea as migrant workers. At present, more than 10,000 Mongolian migrant workers live and work in South Korea. They are usually young, sexually active, and living alone. Their working and living conditions are unstable and looking for friends and sex partners is not easy in a new environment. Given these circumstances, these workers are a vulnerable population for HIV-1 infection. These data clearly demonstrate that education on HIV-1 infection among the migrant workers is not enough and comprehensive actions for the prevention of HIV-1 infection are needed before these workers go abroad.
In conclusion, our study identified a hot spot of HIV-1 transmission expanding currently and the potential seed of the epidemic in Mongolia. Comprehensive preventive measures are crucial to keep the rate of HIV-1 infection low in Mongolia. Our study provided clues for effective strategic actions for HIV-1 prevention.
Footnotes
Acknowledgments
This study was supported by grants from the National Center for Global Health and Medicine (H20-04-R) from the Ministry of Health, Labour, and Welfare of Japan and from the Global Center of Excellence Program (Global Education and Research Center Aiming at the Control of AIDS) from the Ministry of Education, Science, Sports, and Culture of Japan. The authors would like to thank all the doctors and assistant nurses of the AIDS/STI Department NCCD, Mongolia, for their roles in enrolling study participants and collecting blood samples. We are also grateful to all participants who contributed to the study.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.