
Abstract
Human T cell lymphotropic virus type 2 (HTLV-2) infection is endemic in the American Indian population and Pygmy tribes in Africa. Nevertheless, HTLV-2 infection has been predominantly detected in U.S. and European injecting drug users (IDU). Noteworthy is that the HTLV-2a subtype is the main circulating variant in North America and Eastern Europe whereas the HTLV-2b subtype is mainly found in Western Europe, particularly in Italy and Spain where coinfection with HIV-1 is frequent. Twelve Spanish subjects infected with HTLV-2 were recruited for the study. All of them were IDUs coinfected with HIV-1. Molecular epidemiology was done by sequencing the LTR, env, and tax regions and by generating phylogenetic trees. The present study showed that all the sequences belonged to the HTLV-2b subtype and were closely related to other Spanish and Portuguese reported sequences, clearly differentiated from those belonging to the HTLV-2a subtype from Eastern Europe. Therefore, infection with HTLV-2b remains prevalent in Spain based on previous studies.
T
Molecular epidemiological studies have distinguished three main HTLV-2 subtypes geographically dispersed. Subtype HTLV-2a is the main circulating variant in North America and Eastern Europe, while subtype HTLV-2b can be found in native Central and South American Indians as well as in Western Europe, particularly in Italy and Spain 5,6 where coinfection with HIV-1 is very frequent. Some Brazilian isolates clustered within subtype HTLV-2a but diverged by coding a 25 amino acid longer Tax protein. This diversity conceived as a molecular variant of HTLV-2a 7 was considered by some to indicate another subtype (HTLV-2c). 8 Finally, subtype HTLV-2d has been detected among African tribes. 9 Data from the Spanish HTLV national registry showed that a total of 717 HTLV-2-infected individuals had been diagnosed in Spain up to December 2009. 10 Despite of the continuing immigration from HTLV-2 endemic areas, most of the newly infected cases reported in Spain were native former drug users coinfected with HIV-1. 10
In the present study, 12 HTLV-2-infected Spanish individuals (58% male) with a history of intravenous drug use and coinfected with HIV-1 were phylogenetically analyzed through three viral genes and compared with other reported isolates. The individuals analyzed were diagnosed between 2003 and 2009. All the selected samples were obtained from epidemiologically unlinked individuals from the Hospital Ramon y Cajal, located in Madrid, Spain.
Plasma samples from these individuals showed a complete positive HTLV western blot (WB) pattern (Bioblot HTLV, Genelabs, Singapore) with the exception of three samples that showed an indeterminate WB pattern, according to HTLV European Research Network criteria. 11 These indeterminate samples were later confirmed by HTLV-2-specific PCR. DNA was extracted from cryopreserved peripheral blood mononuclear cells (PBMCs) using the QIAamp DNA kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's protocol. Partial LTR, env, and tax genes were amplified by nested polymerase chain reaction (PCR).
All reaction mixtures were performed in a total volume of 25 μl containing PCR Buffer II (AccuPrime Taq High Fidelity, Bioline, London, UK), 0.5 U Taq polymerase (AccuPrime Taq High Fidelity, Bioline), 200 ng of genomic DNA, and 20 pmol of each primer, HT2-1 (TAAAGGCTCTGACGTCTCC-3′) and HT2-2 (GCAGCAAGGGCTAGGGCT-3′) for LTR, Ev-2 (GTTCCAATAGCAGTGAGCCTTGT-3′) and Ev-3 (AAAGCTGCATGCCCAAGAC-3′) for the env gene, and Tx-3 (CTGGTCTCCTAACGGCAATCTC-3′) and Tx-4 (CAAGTAAAGGCTCTGACGTCT-3′) for the tax gene. Thirty-five cycles of denaturation at 94°C for 15 s, annealing at 58°C for 15 s, and extension at 68°C for 20 s were used. After the first PCR reaction, 2 μl of the amplified products was used for a second PCR run in the same amplification conditions. Primer HT2-1 (TAAAGGCTCTGACGTCTCC-3′) and HT2-4 (AACGAAACCTCAACGCCGCC-3′), Ev-3 (AAAGCTGCATGCCCAAGAC-3′) and Ev-4 (ATTGCTACCAACCTCGCCTAC-3′), and Tx-3 (CTGGTCTCCTAACGGCAATCTC-3′) and Tx-6 (CAGACCGTCTCACACAAACAATC-3′) were used for the amplification of 390 bp (nt 153–541) of the LTR fragment, 777 bp (nt 5180–5956) of the env region, and 993 bp (nt 7213–8205) for the tax region, respectively. The sequence positions are listed according to the Mo isolate (GenBank accession number M10060).
Purified PCR products were sequenced in a final volume of 15 μl by using the ABI Big Dye Terminator v3.1 cycle sequencing reaction kit (Applied Biosystems, Foster City, CA) and processed with the ABI 310 Genetic Analyzer (Applied Biosystem). Sequences were edited using SeqMan II Software version 5.0.1 (DNASTAR, Madison, WI) and aligned with MEGA software v4.0.2 together with reference sequences of the HTLV-2a subtype (Mo) and HTLV-2b subtype (G12 and NRA). Phylogenetic trees were generated using the unweight pair group method with arithmetic mean (UPGMA) by Clustal W v5.8 and MEGA v4.0.2. The neighbor-joining method was also used to corroborate them. The topology of the trees was supported by 1000 bootstrap replicates. Genetic distance was estimated by Kimura's two-parameter method. Some reported sequences from GenBank listed below were used in this study for comparison purpose, and the simian T cell lymphotropic virus type 2 (STLV-2) was used as the outgroup.
Within the LTR fragment (nt 153–541), the NF-κB binding site was highly conserved when compared to NRA except for the insertion of an A at position 183 in the MA_CGO, MA_DGE, and MA_JSR sequences. Another A was inserted at position 258 in the MA_MSC and MA_SPG sequences. All sequences showed a transition from G to A at position 305 except MA_GRO and MA_CAS, and two contiguous transversions from TC to GG at positions 323 and 324, respectively (data not shown). Similarity values when compared to prototypes NRA (HTLV-2b) and Mo (HTLV-2a) are shown in Table 1.
The splicing site within the beginning of the env fragment was conserved. This site is essential to generate the double-spliced messengers encoding the regulatory tax protein. 9 The nucleotide sequences were well conserved when compared to NRA, with high similarity values (Table 1). A total of 11 point mutations were found at positions 5231, 5358, 5827, 5832, 5945, and 5949 in MA_BMJ, MA_VMM, MA_SPG, MA_CAS, MA_ROL, and MA_FRM sequences, and at positions 5359 and 5413 in all sequences. The remaining three nucleotide point mutations at 5185, 5209, and 5437 were found in the same subject MA_VMM. Six of these mutations were related to an amino acid change in the coded protein at positions 10, 18, 59, 218, 256, and 257 in the MA_VMM, MA_BMJ, MA_MSC, MA_CAS, MA_ROL, and MA_FRM sequences, respectively (Fig. 1A).

Alignment of Env
Almost the complete tax region (nt 7213–8205) was analyzed. The percentage of nucleotide similarity with regard to both Mo and NRA prototypes is shown in Table 1. A total of 10 point mutations were found when compared to the HTLV-2b NRA prototype at positions 7400 in MA_GRO, 7475 in MA_ROL, 7824 and 8045 in MA_CGO, 7915 in MA_VMM, 8007 and 8030 in MA_MSC, 8010 in MA_JSR, and finally 8144 and 8182 in all sequences except in MA_MSC for the last position indicated, respectively. Generally, the amino acid sequences were well conserved, especially the nuclear location signal (NLS), nuclear export signal (NES), and the dimerization domain that remained unchanged for subtype b. 12 Nevertheless, six of the nucleotide mutations generated an amino acid substitution at positions 63, 88, 235, 273, and 278, in MA_GRO, MA_ROL, MA_VMM, MA_MSC, and MA_CGO sequences, respectively. All the sequences shared the amino acid change at position 323 except MA_MSC, which remained intact with regard to NRA prototype (Fig. 1B). The amino acid identity compared to Mo and NRA sequences is also shown in Table 1.
The phylogenetic tree for LTR showed that Spanish samples clustered with other reported sequences from Spanish (BF, AA, JA, DP, 130, 324, RVP, SPAN129, and SPAN130) and Portuguese (PortNn, PortHl, and PortVs) IDUs. Together these formed a subgroup closely related to sequences from Italian IDUs (I-GI, I-OV, I-OG, and I-OV Gu), and more distantly with sequences from American Indian (WYU1 and G12) and African (Gab) sequences. All these sequences clustered in the HTLV-2b subtype sustained by a bootstrap value of 85%. Subtypes 2a and 2d formed other two genetically distant clusters (Fig. 2).
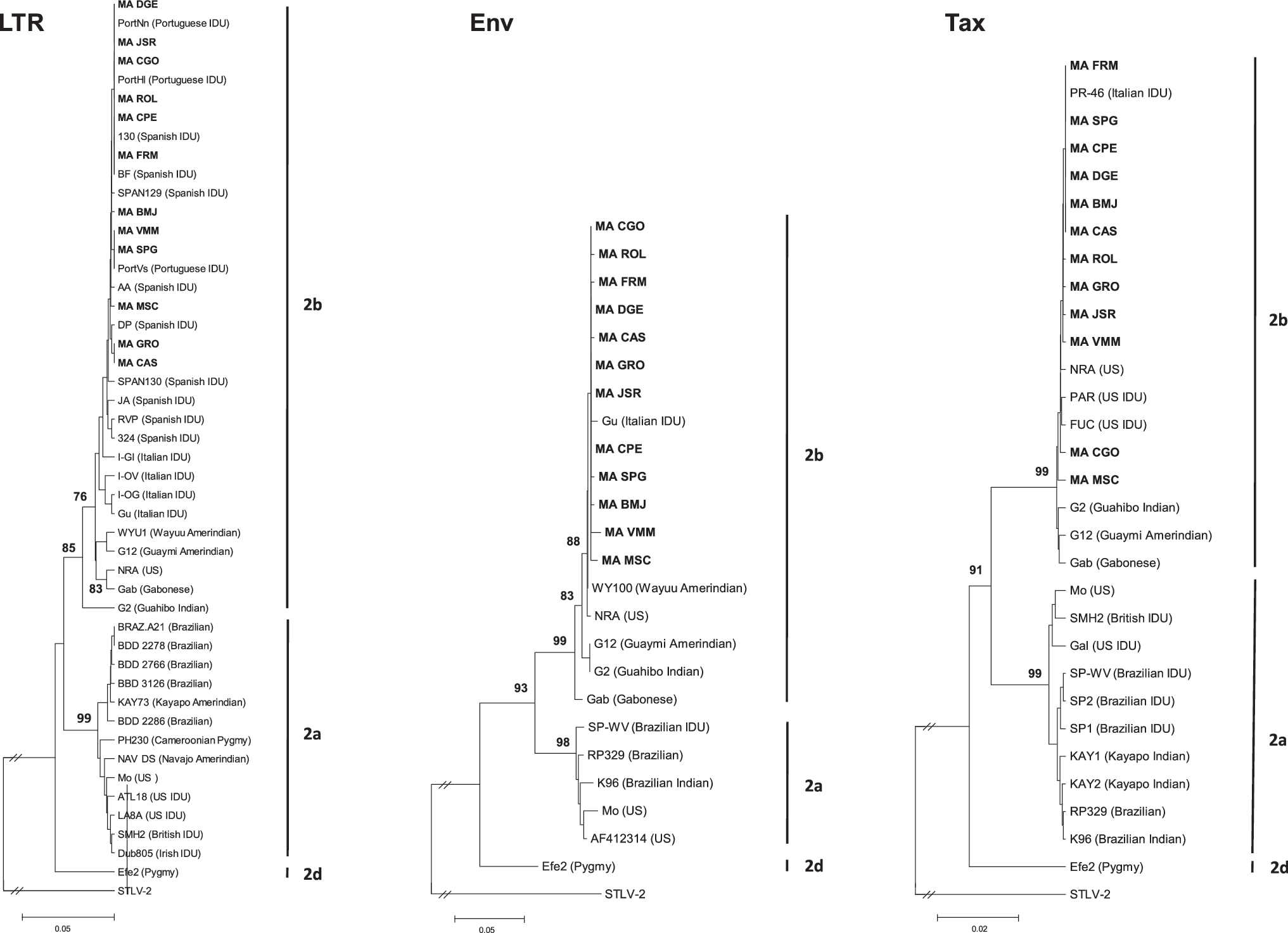
Dendrograms showing the phylogenetic relationship between 320 bp of the LTR region, 777 bp of the env fragment, and 993 bp of the tax region of the HTLV-2 strains. Twelve Spanish sequences are shown in bold and compared to other reported sequences of which the simian T cell lymphotropic virus type 2 (STLV-2) was used as an outgroup for the analysis. Geographic and ethnic origins are shown in parentheses and 1000 bootstrap replicates were performed to support internal branches. The genetic distance bar is also shown.
Similar results were found for env gene analysis where Spanish sequences were highly related to reported sequences from Italian IDUs (I-OV). Together with sequences from Amerindian (WY100, G12, and G2) and African (Gab) individuals, these formed the HTLV-2b subtype supported by a bootstrap value of 99%. The MA_MSC and MA_VMM sequences were the most divergent sequences among the Spanish samples (Fig. 2).
Finally, the analysis of tax region showed three well-differentiated groups for HTLV-2a, HTLV-2b, and HTLV-2d subtypes, supported by high bootstrap values. The 2b subgroup included reported sequences from Italian (PR-46) and American (FUC and PAR) IDUs closely related to the Spanish sequences and the reported sequences from the Gabonese (Gab) and the American Indian prototypes (G12 and G2). Among Spanish samples, MA_CGO and MA_MSC were the most divergent (Fig. 2).
With respect to HTLV-2 infection, most of the cases reported in Spain were native IDUs and coinfected with HIV-1. 13 It has been speculated that HTLV-2 originated from the Amerindians and was introduced to European IDUs in at least two separate periods: one for the 2a subtype in Eastern Europe and another one for the 2b subtype in Western Europe. 14 During the 1980s and 1990s, HTLV-2 spread exponentially among Spanish intravenous drug users because of needle-sharing practices. The prevalence of HTLV-2 infection has been one of the highest in Europe, estimated at 18% among IDUs in prisons and at 1–5% outside the prison setting. 15 In the past decades, many injecting drug users have changed the route of drug usage, thus the number of new injecting drug users has decreased. Therefore, it is estimated that the number of new cases of HTLV-2 infection might be very low. 13
The present study of 12 infected IDUs from Spain coinfected with HIV-1 showed that HTLV-2b remains the prevalent subtype in Spain. They also continue to be closely related to Italian and Portuguese IDUs, clearly differentiated from those belonging to the HTLV-2a cases from Eastern Europe.
Sequence Data
The following GenBank accession numbers for previously published HTLV-2 gene fragments were used in this study: Mo (M10060), G12 (L11456), G2 (AF07965), NRA (L20734), Gab (Y13051), Efe-2 (Y14365), STLV-2 (U90557). For the LTR analysis: NAV.DS (U10257), SMH2 (Y09148), PH230PCAM (Z46838), LA8A (U10256), KAY73 (L42509), BRAZ.A21 (U10253), ATL18 (U10252), Dub805 (AF175467), AA (L7738), DP (L7737), JA (L7739), BF (L77236), SPAN129 (U10265), SPAN130 (U10266), RVP (L77244), 130 (L77242), 324 (L77243), PortHl (AY622977), PortNn (AY622978), PortVs (AY622979), Gu (X89270), I-GI (Y09153), I-OV (Y09155), I-OG (Y09154), WYU1 (U12792), BBD_3126 (FJ911656), BBD_2278 (FJ911644), BBD_2766 (FJ911652), BBD_2286 (FJ911645). For the env analysis: WY100 (S69268), Gu (X89270), SP-WV (AF139382), RP329 (AF326583), K96 (AF326584), AF412314. For the tax analysis: SMH2 (Y09148), Gal (AF292002), SP-WV (AF139382), RP329 (AF326583), K96 (AF326584), KAY1 (U32874), KAY2 (U32875), SP1 (U32873), SP2 (U32872), PR-46 (DQ022075), FUC (U32882), PAR (U32880). Sequences from the samples studied in this work have been submitted to GenBank under the following numbers: LTR: MA_CPE (GU433203), MA_DGE (GU433204), MA_FRM (GU433205), MA_MSC (GU433206), MA_JSR (GU433207), MA_CGO (GU433208), MA_GRO (GU433209), MA_SPG (GU433210), MA_ROL (GU433211), MA_VMM (GU433212), MA_BMJ (GU433213), MA_CAS (GU586121). env: MA_BMJ (GU586109), MA_DGE (GU586110), MA_FRM (GU586111), MA_MSC (GU586112), MA_JSR (GU586113), MA_CGO (GU586114), MA_GRO (GU586115), MA_SPG (GU586116), MA_ROL (GU586117), MA_VMM (GU586118), MA_CPE (GU586119), MA_CAS (GU586120). tax: MA_BMJ (GU591294), MA_CGO (GU591295), MA_CPE (GU591296), MA_DGE (GU591297), MA_FRM (GU591298), MA_GRO (GU591299), MA_JSR (GU591300), MA_MSC (GU591301), MA_ROL (GU591302), MA_SPG (GU591303), MA_VMM (GU591304), MA_CAS (GU591305).
Footnotes
Acknowledgments
We want to thank Maria Pumares and Paloma Martí for their technical support. We thank all the participants of this study for their cooperation. This study was supported in part by Fondo de Investigación Sanitaria (FIS) 07/0070 and Redes Temáticas de Investigación Cooperativa en Salud (RETICS) Red de Investigación en SIDA (RIS) RD/06/0006/0034.
Author Disclosure Statement
No competing financial interests exist.