
Abstract
Broadly neutralizing monoclonal antibody (MAb) 2F5 targets a linear epitope within the highly conserved membrane proximal external region (MPER) of the HIV-1 envelope protein gp41 integral subunit. Prospective vaccine developments warrant efforts currently underway to unveil the mechanistic and structural basis of its mode of action. One open question relates to the putative role that membrane phospholipids might play in the neutralization process. In this work, we establish experimental conditions that allow monitoring 2F5 insertion into lipid bilayers. Then, we compare the abilities of 2F5-based MAb, Fabs, and 2F5-specific antibodies recovered from immunized rabbits to directly penetrate into lipid bilayers and block the lytic activity of MPER-derived peptides. Antibody insertion induced membrane perturbation, which was blocked on interacting with the peptide epitope, thereby suggesting that such phenomenon was primarily mediated by the epitope-binding site. The long, hydrophobic complementarity-determining region (CDR)-H3 loop contributed little to this effect. In contrast, the CDR-H3 loop was required for blocking the lytic activity of MPER-based peptides and viral neutralization. Thus, our results suggest that core epitope binding plus association with lipid bilayers are not in conjunction sufficient to support viral neutralization by 2F5. Moreover, they support a role for the CDR-H3 loop in establishing secondary interactions with lipids and/or gp41 that would block the membrane-perturbing activity of MPER during fusion.
Introduction
M
The conserved MPER domain is enriched in hydrophobic-at-interface aromatic residues, which are proposed to enable its insertion into the virion membrane surface. 21,22 Membrane-inserted MPER might function by generating destabilization of the bilayer architecture during gp41-induced fusion. 21 –27 Its putative functional role in viral entry raises the possibility that binding and blocking of membrane-embedded MPER epitopes may be required for neutralization by antibodies targeting this domain. 26,28 –33 Such a mechanism would entail intimate antibody contacts with the phospholipid bilayer components. Molecular and structural characterization of 2F5 and 4E10 antibodies reveals the presence of long, hydrophobic CDR-H3 loops. 4,28,34 –36 Experimental evidence recently published by various laboratories is overall consistent with those elements playing a key role in neutralization by actually mediating an antibody–membrane association. 37 –41
Evidence of 4E10 and 2F5 polyreactivity with phospholipids (PLs) has also been reported. 29,32,42 –46 The observation that these MAbs may adsorb to PL-coated solid supports has led to the hypothesis that MPER in the context of the membrane may resemble self-antigens similar to those recognized by antibodies identified in a variety of autoimmune diseases. 42 These findings have also provided support for the proposal that preassociation with membranes is required for subsequent 2F5 and 4E10 epitope binding and viral neutralization. 37 Conversely, a direct MAb association with membranes has not been observed in studies employing freely diffusing, electrically neutral unilamellar vesicles with sizes comparable to those of virions, 33 or eukaryotic cells. 47 Under those conditions, the specific binding to an inserted epitope appears to be a prerequisite for MAb–membrane association. Hence, the close proximity to the lipid bilayer surface, attained after epitope engagement, is what apparently enables subsequent antibody partitioning-insertion (see the Discussion below).
Previous data from our laboratory were consistent with the involvement of electrostatic interactions in MAb association with phospholipids. 43 Moreover, our recent results challenge the concept that membrane association plus epitope binding might solely explain the 2F5 neutralizing capacity. 41 With the aim of assessing the contribution of direct 2F5–membrane interactions to MPER blocking and neutralization, we have established conditions that allow monitoring of the direct insertion of this antibody into curvature-free lipid monolayers and large unilamellar vesicles (LUVs) made of pure PL. Our observations indicate that combining membrane-inserting capacity with epitope binding might not be sufficient to recapitulate 2F5 neutralization. In addition, they reveal the existence of a correlation between MPER membrane activity blocking and neutralization, which is dependent on the CDR-H3 loop.
Materials and Methods
Materials
Peptides representing different fragments of the gp41 MPER, 2F5ep (656NEQELLELDKWASLWN671), 2F5ep-Cys (656NEQELLELDKWASLWN671C), 2F5preTM (656NEQELLELDKWASLWNWFNITNWLWYIK683), and 2F5preTM(9,10)Ala (656NEQELLEL-664AA665-WASLWNWFNITNWLWYIK683) were synthesized by solid-phase synthesis using Fmoc chemistry and purified by HPLC at the Proteomics Unit of the University Pompeu-Fabra (Barcelona, Spain). Overlapping HIV-1 consensus subtype B env (15-mer) peptides 8923 to 8928 (contributed by Anaspec, Inc.) and HIV-1 gp120 MAb F105 (contributed by M. Posner and L. Cavacini) were obtained from the NIH AIDS Research and Reference Reagent Program. MAb 2F5 was supplied by Polymun Scientific GmbH (Vienna, Austria). 2F5 Fab expression, purification, and mutagenesis have been described in detail in a preceding article. 41 Phosphatidylglycerol (PG), cardiolipin (CL), phosphatidylinositol (PI), phosphatidylserine (PS), phosphatidylcholine (PC), phosphatidylethanolamine (PE), sphingomyelin (SPM), and cholesterol (Chol) were purchased from Avanti Polar Lipids (Birmingham, AL). The 8-aminonaphthalene-1,3,6-trisulfonic acid sodium salt (ANTS) and p-xylenebis(pyridinium)bromide (DPX) fluorescent probes were from Molecular Probes (Junction City, OR).
Monolayer penetration
Surface pressure was determined following the Wilhelmy method in a fixed-area circular trough (μ Trough S system, Kibron, Helsinki). The measurements were taken with constant stirring in an aqueous phase consisting of 1 ml 5 mM HEPES and 100 mM NaCl (pH 7.4). Lipid mixtures dissolved in chloroform were spread over the surface at the initial surface pressure (π0) of ca. 22–25 mN/m. Antibodies were injected into the subphase with a Hamilton microsyringe and the surface pressure changes were continuously recorded via a computer interface.
Production of vesicles
LUVs were prepared following the extrusion method of Hope et al. 48 Phospholipids dissolved in chloroform were dried under an N2 stream. Traces of organic solvent were removed by overnight vacuum pumping. Subsequently, the dried lipid films were dispersed in 5 mM HEPES, 100 mM NaCl (pH 7.4) buffer, and subjected to 10 freeze–thaw cycles prior to extrusion 10 times through two stacked polycarbonate membranes (Nuclepore, Inc., Pleasanton, CA). The size distributions of the vesicles were determined using a Malvern Zeta-Sizer Nano ZS instrument (Malvern Instruments, Malvern, UK). Extrusion through membranes with a nominal pore size of 0.1 μm produced PG LUV with mean diameters of ca. 110 nm. Lipid concentrations of liposome suspensions were determined by phosphate analysis.
Leakage assay
The release of aqueous vesicular contents into the medium was monitored by the ANTS/DPX assay. 49 LUVs containing 12.5 mM ANTS, 45 mM DPX, 20 mM NaCl, and 5 mM HEPES were obtained by separating the unencapsulated material by gel filtration on a Sephadex G-75 column eluted with 5 mM HEPES and 100 mM NaCl (pH 7.4). Osmolarities were adjusted to 200 mosm in a cryoscopic osmometer (Osmomat 030, Gonotec, Berlin, Germany). Fluorescence measurements were performed in a thermostatized cell with constant stirring, after setting ANTS emission at 520 nm and excitation at 355 nm. A cutoff filter (470 nm) was placed between the sample and the emission monochromator. The 0% leakage value corresponded to the fluorescence of the vesicles at time zero whereas 100% leakage was defined as the fluorescence value obtained after addition of Triton X-100 (0.5%, v/v).
Rabbit immunization and antibody isolation
For immunization, 2F5preTM was added at a final peptide-to-lipid ratio of 1:50 (mol:mol) to a stirring solution of freeze–thaw PG vesicles dispersed in phosphate-buffered saline (PBS). After incubation for 30 min, the samples were lyophilized. New Zealand White rabbits were inoculated intradermally at multiple sites on day 0 with 1 ml of sample reconstituted in pure water, which contained 0.5 mg peptide supplemented with 1.25 mg of muramyl dipeptide (Sigma-Aldrich, St. Louis, MO). One milliliter of the reconstituted liposome formulation containing 0.3 mg peptide was used for subsequent boosting injection on day 15 (0.3 mg peptide), while 0.2 mg of liposomal peptide was injected on days 30, 45, and 60. 2F5 epitope-specific antibodies were recovered from sera through affinity purification. To that end, 2F5epCys was immobilized to a beaded agarose support using a Sulfolink Immobilization Kit for Peptides (Thermo Scientific, Rockford, IL) following the manufacturer's instructions. The remaining nonspecific binding sites in columns were blocked adding
Every analyzed serum was loaded on the columns after diluting and filtering it to remove the particulate material. They were let to flow through the columns four times allowing the binding of all the antibodies present in the serum that recognize specifically the immobilized peptide. After washing the columns with at least 10 bed volumes of 500 mM NaCl containing buffer to dispose of nonspecifically bound antibodies and serum proteins, the specific antibodies (Bound) were eluted using 100 mM glycine buffer at pH 2.5. The fraction that is not recovered using acidic pH was eluted using freshly made 100 mM triethylamine buffer at pH 11.5. This latter fraction was mostly devoid of 2F5-specific antibodies. IgGs present in the unbound, eluted fractions (Eluted) were isolated using Protein-G-Sepharose HiTrap Protein G HP columns (Amersham Biosciences Europe GmBh, Freiburg, Germany), and subsequently used as negative controls for specific epitope recognition.
Neutralization assays
Pseudoviruses were produced by transfection of human kidney HEK293T cells with the full-length env clone pHXB2-env (AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, contributed by K. Page and D. Littman) using calcium phosphate, together with vectors pWXLP-GFP and pCMV8.91, encoding, respectively, a green fluorescent protein and an env-deficient HIV-1 genome (kindly provided by Dr. Patricia Villace, CSIC, Madrid). After 24 h, the medium was replaced with Optimem-Glutamax II (Invitrogen Ltd, Paisley, UK) without serum. Two days after transfection, the pseudovirus particles were harvested, passed through 0.45-μm pore sterile filters (Millex HV, Millipore NV, Brussels, Belgium), and finally concentrated by ultracentrifugation in a sucrose gradient. Neutralization was determined using TZM-bl target cells. Samples were set up in duplicate in 96-well plates, and incubated for 1 h at 37°C with a 10–15% tissue culture infectious dose of pseudovirus. After Fab-pseudovirus coincubation, 10,000 target cells were added in the presence of 15 μg/ml DEAE-dextran (Sigma-Aldrich, St. Louis, MO). Neutralization levels after 72 h were inferred from the reduction in the number of GFP-positive cells as determined by flow cytometry using a BD FACSCalibur Flow Cytometer (Becton Dickinson Immunocytometry Systems, Mountain View, CA).
Results
MAb2F5 is surface active, inserts into lipid monolayers, and permeabilizes vesicles made of anionic phospholipids
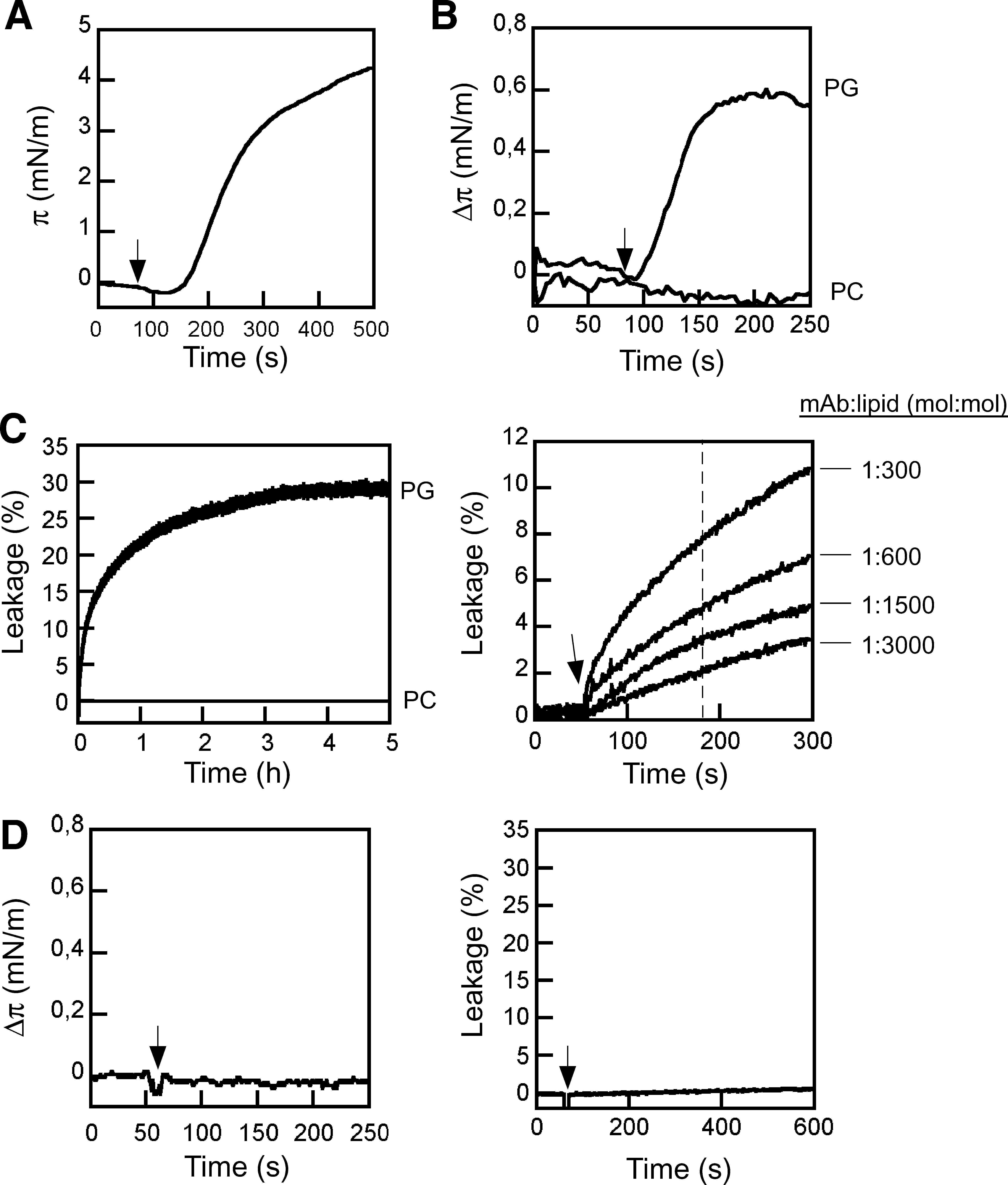
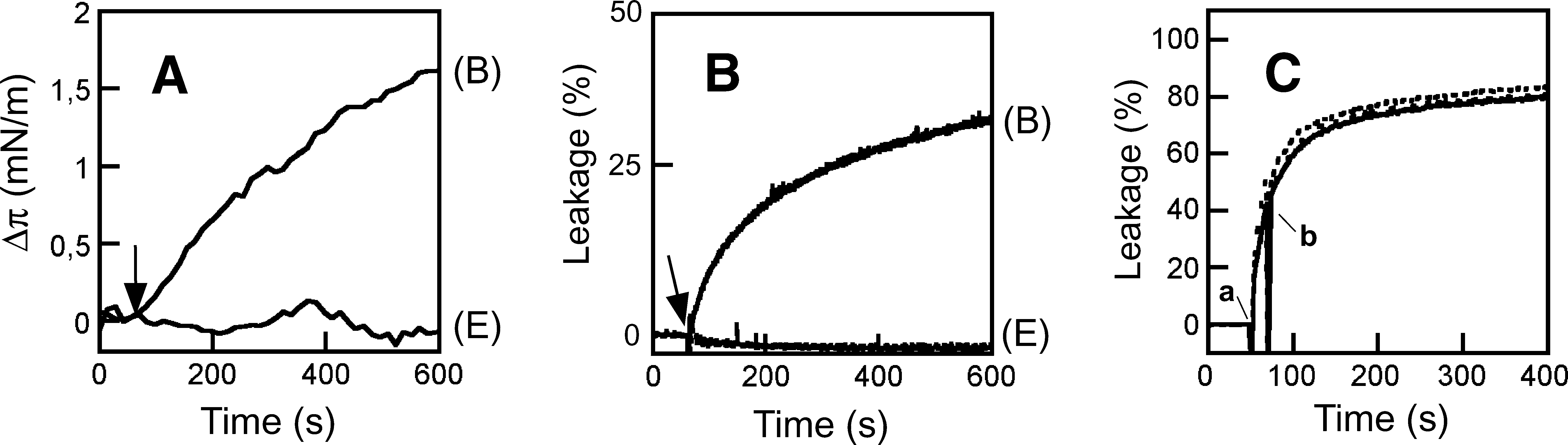
The lipid monolayer systems provide homogeneous membrane surfaces, 50,51 i.e., devoid of the packing defects that may arise in other membrane model systems upon dispersion of dried lipids in water solution, or after immobilization of the resulting bilayers onto solid supports. Since lipid dispersions have been used in the past to produce results that sustain 2F5 polyreactivity to lipids, monolayers constitute a suitable, additional model for contrasting the relevance of this phenomenon. Thus, we made use of lipid monolayer-based experiments to obtain direct proof of MAb2F5 insertion into membranes. Surface activity at the air/water interface was first determined to assess the amphipathic character of the water-soluble MAb molecules. The adsorption of the 2F5 antibody at the air/water interface was monitored by following the increase of surface pressure as a function of time (Fig. 1A). MAb addition to the aqueous subphase resulted in an increase of pressure that reached a saturation π∞ value of ca. 5 mN/m. The surface activity at the air/water interface is determined primarily by the exposed nonpolar residues. 51 Thus, results displayed in Fig. 1A give support to the possibility that the hydrophobic residues exposed to solvent might drive efficient antibody incorporation into a hydrophobic phase.
Membrane activity of MAb2F5.
The subsequent changes induced in lipid monolayer surface pressure confirmed the penetration into membranes composed of anionic PLs (Fig. 1B). Injection into the subphase of 35 μg/ml of 2F5 induced an increase of surface pressure of PG monolayers initially compressed at ca. 22 mN/m. Control experiments indicated that injection of the same amount of an irrelevant antibody (anti-gp120 F105) did not alter the monolayer surface pressure (Fig. 1D, left). By comparison, the 2F5 MAb did not induce any appreciable pressure increase in electrically neutral PC monolayers. Table 1 illustrates the dependency of MAb2F5 insertion on the lipid composition. These monolayer penetration results, in combination with the adsorption to the air/water interface, were consistent with a significant capacity of 2F5 for inserting into negatively charged membranes, and therefore suggested the involvement of hydrophobic and electrostatic interactions in the process.
Surface pressure increases with initial monolayer pressure fixed at ca. 22 mN/m: ++, 1–10 mN/m increases; +, 0–1 mN/m; −, no increase.
CL, cardiolipin; PG, phosphatidylglycerol; PI, phosphatidylinositol; PC, phosphatidylcholine.
In many instances, insertion into the bilayer compromises the permeability barrier of lipid vesicles. In search of additional evidence for intimate interaction with membranes, we next tested the potential capacity of 2F5 to induce vesicle permeabilization. LUVs with a mean diameter of ca. 100 nm were selected as bilayer models to confer a curvature comparable to that of the viral envelope. Figure 1C (left panel) shows the time course of ANTS leakage from PG vesicles incubated with 2F5 MAb. 2F5-induced aqueous contents spilling of PG LUV reached a plateau after several hours. Again, addition of the irrelevant anti-gp120 F105 antibody to the vesicle suspension did not alter the permeability (Fig. 1D, right). Saturation of the process with time is consistent with a permeabilization process mediated by structures irreversibly attached to the lipid bilayers, i.e., not interchanging between the different vesicles in the samples. By analogy to pore-forming proteins, partial release in the presence of an excess of antibody might also reflect the requirement of a minimal number of antibodies per vesicle for induction of bilayer destabilization.
In contrast, no ANTS was released from PC vesicles treated with 2F5 antibody, thereby indicating that the permeability barrier of these vesicles remained intact upon MAb addition. 2F5 effectiveness as a lytic agent could be evidenced upon PG LUV incubation with different MAb doses (right panel). Protein-to-lipid mole ratios of ca. 1:3000 were sufficient to induce significant levels of vesicle permeabilization. Occurrence of leakage at such low protein doses confirms that lysis was not a mere consequence of adding massive amounts of antibody to the bounding vesicle membrane, but an intrinsic property of each antibody molecule inserted into the PL bilayer.
Table 2 summarizes the lipid compositions sustaining the lytic effect of the antibody. Among the anionic lipids tested, MAb2F5 permeabilized PI and PG vesicles to a similar extent, but did not destabilize vesicles made of CL. This result was unexpected in view of the described 2F5 polyreactivity to this lipid 42,43 and the previous lipid monolayer insertion data (Table 1). The headgroup spacing in CL, a PL with an effective cone shape, 52 might facilitate the accommodation of 2F5 components into the membrane without altering the bilayer permeability barrier. This would also explain the higher 2F5 insertion levels into monolayers made of this lipid but the absence of lytic effects measured in vesicles. In contrast, accession from the water phase to the hydrocarbon core of cylindrical PG and PI bilayers might be more restricted, while inserted 2F5 moieties might induce more disruptive effects on the lipid packing. Electrically neutral PL mixtures or viral-like compositions did not sustain the permeabilizing activity of the antibody. Moreover, inclusion of cholesterol, one of the main lipidic components of the viral membrane, 53,54 inhibited the leakage process observed in PG vesicles.
Mole ratios in zwitterionic and viral-like mixtures were as follows: PC:Chol (2:1); PC:PE:SPM:PS (0.5:1:1:0.5); PC:PE:SPM:PS:Chol (0.5:1:1:0.5:2).
Mole ratios (mol:mol).
Referred to the maximum attained after 5 min of incubation of PG pure vesicles with 25 μg/ml of MAb.
2F5 membrane activity maps primarily to the antigen-binding site
From the previous data it can be concluded that the lytic activity represents an intrinsic property of the 2F5 antibody, which is not functional in the context of the viral membrane. Nonetheless, surface-pressure increase and vesicle-contents leakage constitute suitable assays to monitor direct antibody insertion into membranes (i.e., operating in the absence of a membrane-bound peptide epitope). Thus, these assays were subsequently applied to the identification of the antibody components that are involved in driving the antibody–membrane interaction, which are also predicted to mediate lipid reactivity. Results displayed in Fig. 2 suggest that the PG permeabilization process occurs following the electrostatic interaction of the 2F5 antigen-binding site with the negatively charged lipid bilayer surface.
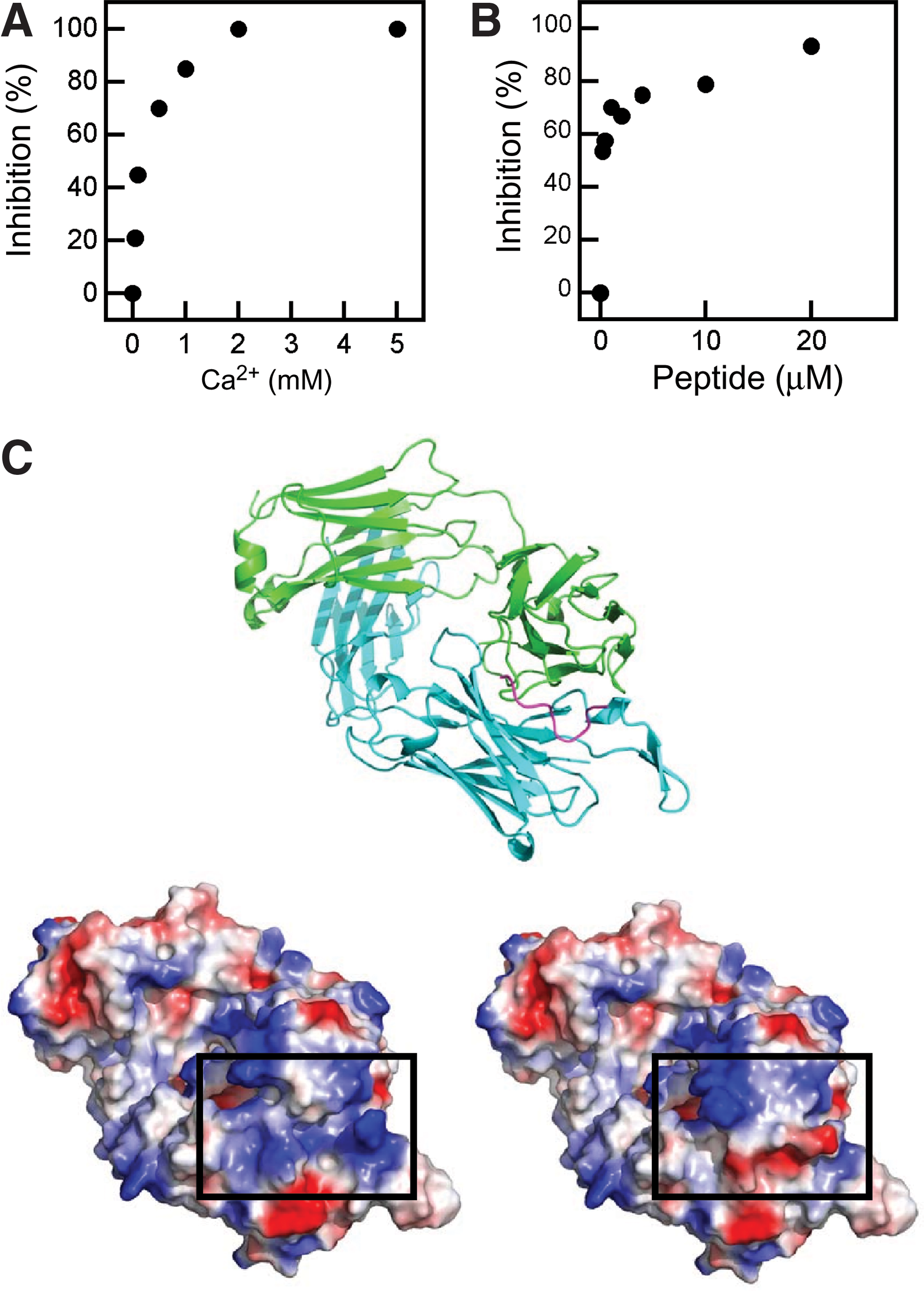
Blocking of 2F5-induced permeabilization by calcium and peptide epitope.
Calcium has been shown to interact with the PG polar headgroup and neutralize the net negative surface charge of lipid bilayers made of this phospholipid. 55 Addition of this cation inhibited the leakage process (Fig. 2A). Similarly, preincubation in solution of the MAb with the 656NEQELLELDKWASLWN671 peptide epitope could effectively block vesicle permeabilization (Fig. 2B), consistent with the involvement in the process of paratope residues engaged in epitope recognition. Of note, the peptide used to carry out these experiments was soluble (i.e., it did not partition from water into membranes) and therefore devoid of any lytic activity. 33
Together, these findings point to the involvement of positively charged 2F5 paratope surfaces in the insertion-leakage process. Structures of the 2F5 Fab displayed in Fig. 2C support this hypothesis. Paratope residues surrounding the linear determinant-binding crevice display a positively charged surface (box in the bottom-left panel). Binding of the peptide–epitope confers an opposite, net negative surface charge to the region (box in the bottom-right panel). Thus, maintenance of net positive and net negative charges, at the paratope and the lipid bilayer surfaces, respectively, appears to be required for driving the MAb insertion-permeabilization phenomenon.
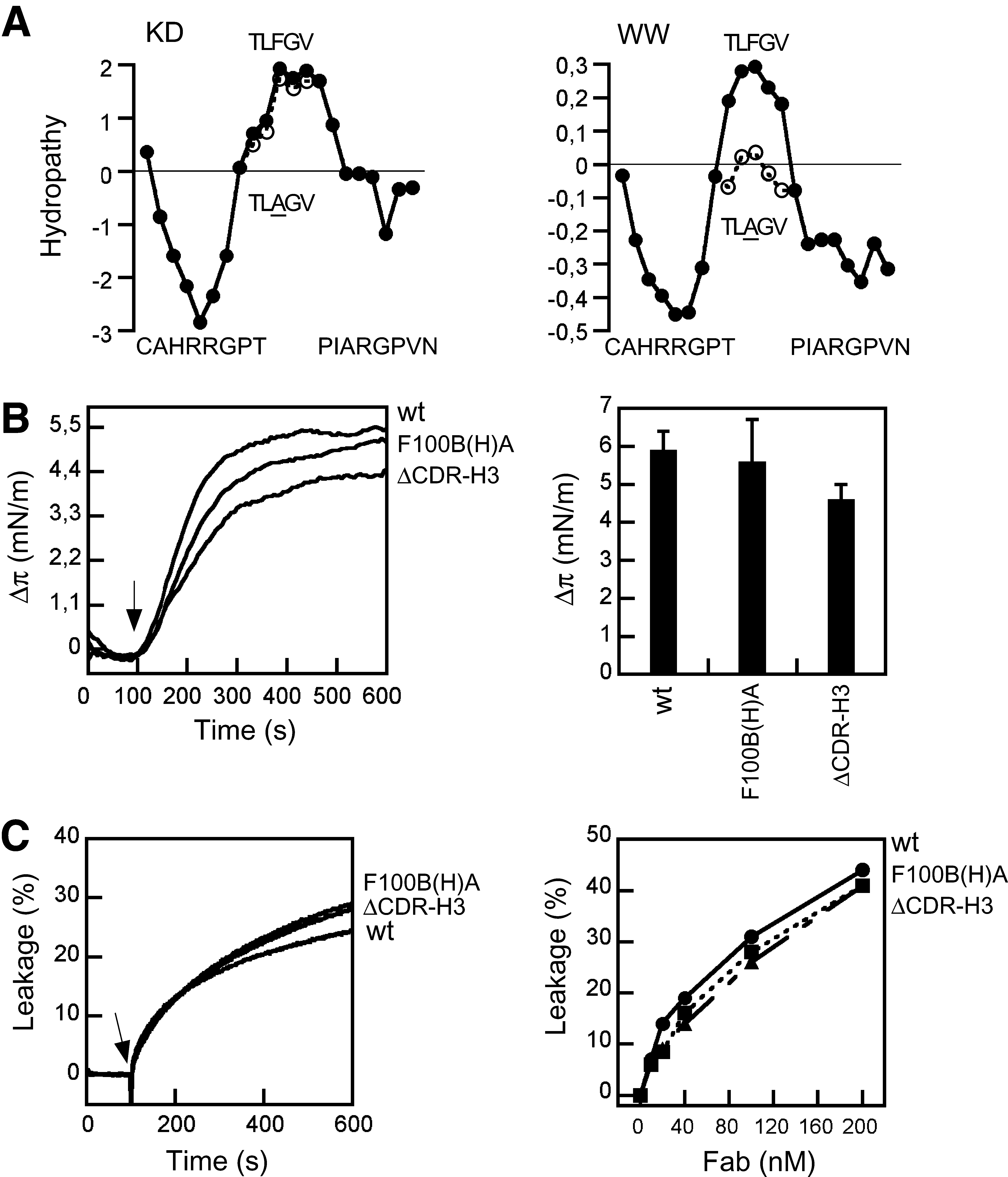
To explore the contribution of the 2F5 CDR-H3 loop to these processes, we next assessed in the PG system the recombinant Fab2F5 variants described in our previous work 41 (Fig. 3). In brief, based on the recombinant wt Fab2F5, two CDR-H3 loop mutants were generated. In the F100B(H)A mutant, the apical Phe100B(H) of the CDR-H3 was changed to an alanine residue. As disclosed in Fig. 3A, the removal of the aromatic phenyl moiety at the apex of the CDR-H3 loop caused the ablation of a positive peak from the Wimley–White hydropathy plot. In contrast, the hydropathy plot was not appreciably affected when the Kyte–Doolittle hydrophobicity scale was applied. Thus, the Phe100B(H)Ala substitution is predicted to interfere specifically with the favorable 2F5 Fab–membrane interface interaction. In the ΔCDR-H3 mutant, the CDR-H3 sequence of 100TLFGVPI100F was replaced by a single Ser-Gly dipeptide, which rendered a Fab essentially devoid of the hydrophobic loop apex. Previous structural studies had revealed that this element did not establish contact with bound peptide epitope residues. 28,36 Consistent with these observations, the loop apex residues did not contribute to 656NEQELLELDKWASLWN671 peptide-epitope binding by the 2F5 Fab. 41
2F5 Fab insertion into lipid monolayers and induced bilayer permeabilization.
Unexpectedly, wild-type (wt), F100B(H)A, and ΔCDR-H3 2F5 Fabs disclosed comparable capacities for inserting into PG monolayers (Fig. 3B) and for inducing PG LUV permeabilization (Fig. 3C). Thus, a 2F5 Fab species with the hydrophobic loop apex ablated was capable of inserting into and permeabilize membranes. Together with the MAb results described previously in Fig. 2, this observation implies that the paratope elements directly in contact with the peptide-epitope, and not the hydrophobic loop apex, are the ones responsible for driving the membrane interactions described in the present work.
MPER membrane-activity blocking by 2F5 antibody is dependent on the CDR-H3 loop and correlates with neutralizing activity
The PG vesicles also provided a suitable system to analyze the contribution of 2F5 paratope elements to membrane-inserted MPER recognition and blocking. Incubation of MPER-based peptides with lipid vesicles results in the release of the aqueous contents to the medium, 22,33 a phenomenon that is proposed to reflect the membrane-perturbing activity of this gp41 domain during fusion. 24 Addition of MAb2F5 to a vesicle suspension arrested ongoing leakage induced by the MPER-based 2F5preTM peptide (Fig. 4A, left), and this inhibition process was dependent on specific 2F5 epitope recognition (Fig. 4A, right). Results shown in Fig. 4B demonstrate that approximately equimolar paratope-to-peptide epitope mole ratios were required to attain total inhibition of leakage.

Inhibition of 2F5preTM-induced membrane permeabilization by MAb2F5.
The flat curve obtained upon antibody addition in Fig. 4A, left (solid line) is also consistent with inhibition of MAb2F5-induced leakage under those conditions. The finding that an antibody that induces leakage by itself was capable of arresting vesicle permeabilization induced by another agent seemed counterintuitive at first glance. However, this observation actually provides additional evidence for the involvement of the epitope-binding site in direct vesicle permeabilization (Fig. 2C). Binding to membrane-inserted 2F5preTM peptide-epitope inhibits both leakage induced directly by the antibody through the antigen-binding site (Fig. 1C) and peptide-induced membrane permeabilization (Fig. 4A). Thus, even though the antibody is endowed with an intrinsic capacity of permeabilizing membranes through the antigen-binding site, engagement of this element in epitope binding results in overall blocking of MPER lytic activity. In conclusion, the antibody–peptide complex formed at the membrane surface would be devoid of the lytic character of its components. 33
To determine the role played by the CDR-H3 loop in the process, the ability of the 2F5 Fab variants to block MPER membrane activity was assessed next (Fig. 5). Similarly to the MAb case, preincubation with soluble 2F5ep peptide-epitope inhibited the direct leakage induced by the 2F5 Fabs (insets). This is also consistent with the comparable affinity of the mutants [F100B(H)A and ΔCDR-H3] and the wt 2F5 Fab for binding the peptide epitope. 41 Also resembling the MAb activity, addition of wt Fab efficiently arrested ongoing leakage induced by the 2F5preTM peptide (Fig. 5, left panel). By comparison, addition of F100B(H)A resulted in a reduced leakage inhibition (Fig. 5, center), while addition of the ΔCDR-H3 Fab did not substantially affect the process (Fig. 5, right). Thus, binding of 2F5 Fab to membrane-inserted 2F5preTM resulted in blocking of the vesicle permeabilization processes induced by both Fab and peptide, but these inhibitory activities appeared to be dependent on different paratope elements: the epitope-binding site and the hydrophobic CDR-H3 loop, respectively.
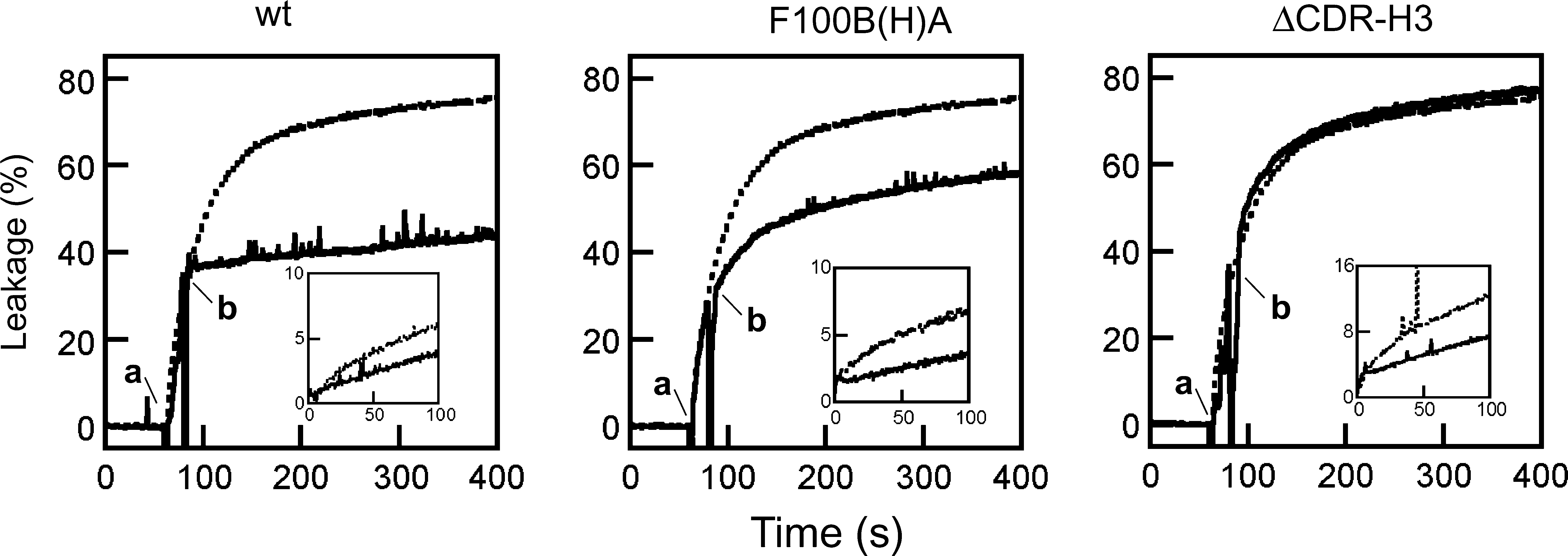
Inhibition of 2F5preTM-induced membrane permeabilization by 2F5 Fabs. Effect of Fab addition to the ongoing leakage. PG vesicle samples (100 μM lipid) were treated with 0.1 μM 2F5preTM and subsequently supplemented with 5 (wt and F100(H)A) or 10 (ΔCDR-H3) μg/ml of 2F5 Fabs (addition times of peptide and antibody are indicated by “a” and “b,” respectively). The dotted traces correspond to the leakage kinetics in the absence of antibody. Insets: Fab-induced direct leakage (dotted lines) was inhibited by preincubation with soluble 2F5ep peptide-epitope (1 μM) (continuous lines).
Finally, the results displayed in Fig. 6 illustrate the correlation existing between the loop-dependent processes of MPER membrane activity blocking and viral neutralization (Fig. 6, left and right panels, respectively). Both inhibitory activities diminished after reducing interfacial loop hydrophobicity, and disappeared upon hydrophobic apex ablation. Interestingly, approximately equimolar paratope:peptide ratios were required for total blocking of MPER-induced leakage by the wt Fab, an observation that underscores the similarity between Fab and MAb inhibitory mechanisms (see Fig. 4B).
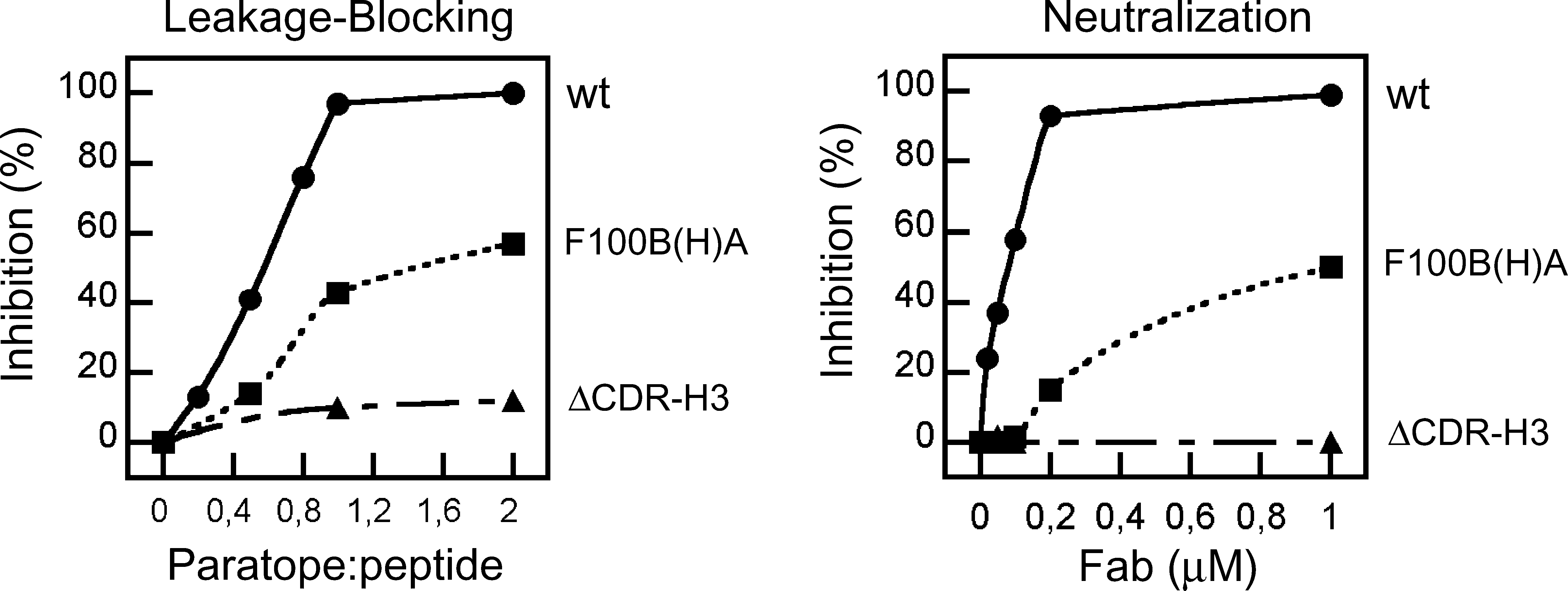
Fab-mediated inhibition of the membrane proximal external region (MPER) membrane-restructuring activity and neutralization. Left: Dose dependency of 2F5preTM-induced leakage inhibition by 2F5 Fabs. Inhibition percentages, calculated from the extents of leakage after 120 s, were plotted as a function of paratope:peptide mole ratio. Right: Neutralizing activity of the 2F5 Fabs measured in a pseudovirus infection assay.
Antibodies specific for the 2F5 epitope recovered in rabbits insert into membranes but do not block MPER membrane activity and are not neutralizing
The previous observations suggest that inhibition of MPER membrane-restructuring activity and viral neutralization may be loop-dependent, interrelated 2F5 properties. They also reveal that 2F5 species devoid of the neutralizing function are still capable of binding to the linear epitope and inserting into lipid bilayers. Hence, recovering the paratope-dependent capacities for membrane insertion and gp41 2F5 epitope recognition might not suffice to recapitulate through vaccination the neutralizing activities displayed by 2F5 MAb and Fabs. This idea is supported by the rabbit immunization results displayed in Figs. 7 and 8. Enzyme-linked immunosorbent assays (ELISA) on sera recovered from rabbits immunized with PG-2F5preTM vesicles revealed a weak, but significant immunogenic response over preimmune sera (not shown). Antibodies specific for the 2F5 epitope could be recovered from these sera after affinity purification (Fig. 7A), but not from preimmune sera (not shown). Titration on ELISA plates with immobilized 2F5preTM also showed that antibodies specific for the 2F5 epitope sequence were absent from the eluted fractions. ELISA mapping subsequently confirmed the antibody specificity for the gp41 region spanning the 2F5 epitope (Fig. 7B and Table 3). Further characterization indicated that changing 664DK665 residues to alanine significantly reduced the amount of bound antibody, consistent with the existence of an anti-2F5ep antibody fraction specific for the correct 2F5 epitope sequence (Fig. 7C). However, these 2F5ep-specific antibodies showed no neutralizing activity in a pseudovirus assay (Fig. 7D).
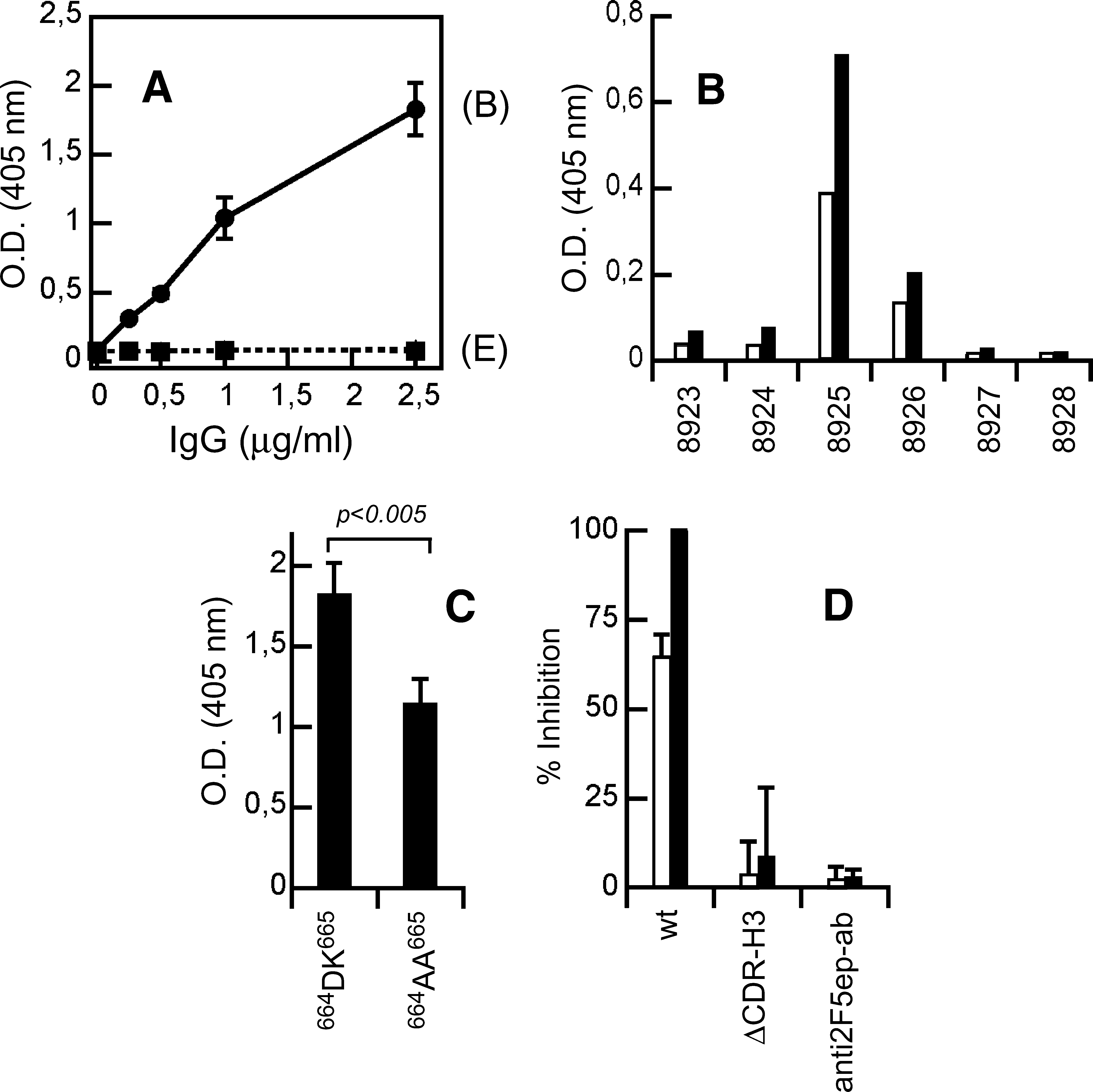
Immunogenicity of PG:2F5preTM mixtures in rabbits.
Membrane interactions of anti-2F5ep antibodies recovered from rabbits.
Numbers as provided in the “NIH AIDS Research and Reference Reagent Program” catalog.
Based on HIV-1 consensus subtype B Env.
The prevailing view indicates that the membrane-inserting capacity of 2F5 is required for neutralization. Thus, the induced polyclonal responses in rabbits might conceivably be devoid of this capacity. Alternatively, the triggered humoral response could be at least in part reminiscent of the ΔCDR-H3 Fab, i.e., be consistent of antibodies endowed with the capacity for inserting into membranes, but devoid of the 2F5 neutralizing activity. To contrast these possibilities, we next assayed the isolated anti-2F5ep antibodies in lipid monolayers and vesicles (Fig. 8). The lipid monolayer results confirmed the capacity of the affinity-purified antibodies for inserting into membranes, while the fraction eluted from columns used as a negative control did not alter the surface pressure (Fig. 8A). In line with these observations, the antibodies specific for the 2F5 epitope, but not the eluted fractions, were capable of permeabilizing PG vesicles directly (Fig. 8B). Finally, anti-2F5ep antibodies were not capable of arresting ongoing leakage induced by 2F5preTM (Fig. 8C). Overall, these results would be consistent with the recovery of ΔCDR-H3-like responses after rabbit immunization with PG-2F5preTM liposome-peptide formulations.
Discussion
The mechanism underlying the broad neutralizing activity of anti-MPER 4E10 and 2F5 antibodies remains elusive. A series of recent publications focused on the involvement of the long, hydrophobic CDR-H3 loop in supporting the process through direct interactions with the viral membrane. 37 –40 In line with the major findings described in those works, we reported that a reduced interfacial hydrophobicity at the loop apex caused the F100B(H)A Fab2F5 mutant to be less efficient in neutralization assays. 41 Moreover, total ablation of this element resulted in a mutant Fab, ΔCDR-H3, devoid of neutralizing activity. Consistent with proper folding of the epitope-binding sites, the mutant and wt Fabs bound to soluble peptide-epitope with comparable affinities. All three Fabs also bound to ELISA plates coated with membrane components, but were unable to appreciably associate with lipid vesicles in solution. However, the Fab mutants showed a decreased affinity for an MPER peptide elongated at the C-terminus; this effect became even more significant when the peptide was bound to membranes. Our results in the present work provide additional experimental evidence to rationalize those findings.
Contribution of 2F5 paratope elements to the interaction with membranes and implications for lipid polyreactivity
As previously discussed, the specific formats of the binding assays may condition the levels of 2F5 antibody reactivity to lipids. 44,56 Thus, PLs applied to ELISA plates may attain distinct physical phases upon uncontrolled hydration, possibly including micelles, bilayers, and inverted hexagonal phases, which might differently condition the availability of sites for antibody binding within exposed surfaces. 46 Even for the lipid bilayers surrounding vesicles, the different surface hydration degrees, the hydrophobic thicknesses, and the material (elastic) properties all might restrict in an interrelated way the packing and surface exposure of different PL molecules for protein binding. 52,57 For instance, in strongly bent small unilamellar vesicles (SUVs) obtained through sonication, the high curvature of the external monolayer increases the polar headgroup spacing and thereby promotes easier access to the hydrocarbon core. 29,43 Vesicles deposited onto solid substrates may also undergo elastic deformations that increase the lipid headgroup spacing, and similarly expose more of the hydrocarbon region for penetration. Thus, MAb insertion may be artificially enabled in stressed vesicular lipid bilayers, under conditions that poorly reflect the physiological state in cell membranes. 47
Unprecedented evidence for MAb2F5 penetration into membranes was obtained using lipid monolayers (Fig. 1B). Since the nature and packing of the constituent PL molecules can be determined without altering the surface curvature, the complexity of vesicular lipid bilayers is greatly reduced in these systems. 50 Moreover, the magnitude of the monolayer surface pressure change induced by a given protein correlates with its capacity for inserting into membranes from the water phase. 51 Consequently, the increase in surface pressure induced by MAb2F5 correlated with a certain capacity of this antibody for inserting into negatively charged lipid bilayers, which was indeed supported by the permeabilization phenomenon observed upon incubation of PG vesicles with MAb2F5 (Fig. 1C).
The observation that under comparable experimental conditions 2F5 did not affect the surface pressure of electrically neutral lipid monolayers (Table 1) was indicative of insertion being primarily limited by the establishment of favorable electrostatic interactions with the membrane surface. Neutralization of the net negative membrane surface charge, either by inclusion in the vesicle composition of electrically neutral PLs or by adding calcium, also inhibited leakage (Table 2 and Fig. 2A). In addition, membrane permeabilization could be blocked by preincubation in solution with a soluble peptide-epitope (Fig. 2B). In concert, this evidence is consistent with the involvement of the positively charged binding site surface in the 2F5 insertion-leakage process, a possibility that is further supported by a comparison between Fab structures with and without bound peptide ligand (Fig. 2C).
A favorable interaction with the membrane is also predicted for the 2F5 CDR-H3 loop (see Fig. 3A). 28,29,40 In principle this long and hydrophobic stretch might insert into the membrane and contribute to the increase in surface pressure and permeabilization. Unexpectedly, neither removal of the affinity for the membrane interface in this element by mutation [F100(H)A substitution] nor ablation of its hydrophobic apex (ΔCDR-H3 deletion) appreciably affected the capacities of 2F5 Fabs for inserting into lipid monolayers (Fig. 3B) or for inducing leakage of aqueous contents from PG vesicles (Fig. 3C).
Thus, attending to the experimental evidence presented here, it can be concluded that 2F5 has a tendency to associate with negatively charged membrane surfaces through the epitope-binding site. This association involves intimate contacts with the membrane matrix that might effectively perturb the lipid bilayer architecture. The observation that insertion-permeabilization is driven through favorable electrostatic interactions also suggests that this MAb would not associate spontaneously with the electrically neutral, plasma membrane surface of normal cells, even if PLs therein were accessible for binding. 44,47,56 Importantly, our data also rule out an essential role for the CDR-H3 loop in these processes, indicating that this element is probably not the main determinant for the previously reported 2F5 lipid polyreactivity (see also below).
Finally, it has been postulated that phosphate-binding subsites exist within the anti-MPER neutralizing antibodies that might engage phospholipid polar headgroup moieties. 39,58 Confirming this prediction, crystallographic data reveal the presence of a potential phosphate-binding subsite at the base of the 2F5 CDR-H3 loop. 36 Thus, the membrane permeabilization effect described here could arise from favorable electrostatic–hydrophobic interactions via the 2F5 epitope-binding site plus specific interactions established with the interfacial phosphate region.
CDR-H3 loop-dependent MPER membrane-activity blocking and implications for the neutralization mechanism
Alam et al. 37 found that 4E10 scFv-s (single chain variable fragments) and recombinant 2F5 IgGs bind to viral lipid mixtures and liposomes immobilized onto solid supports to different extents, depending on the CDR-H3 loop hydrophobicity. Specifically, these authors provided data on a 2F5 IgG mutant bearing the same F100B(H)A substitution analyzed in this work (Fig. 3). In contrast to our observations based on experiments involving Fabs that interact with monolayers and vesicles diffusing in solution, this IgG mutant bound to immobilized liposomes less extensively than the wild-type antibody. However, consistent with our findings, 41 the F100B(H)A IgG was also deficient in neutralization. Based on these observations, Alam and co-workers proposed a two-step mechanism for neutralization according to which 2F5 and 4E10 antibodies first attach to the viral membrane through the CDR-H3 loops. This reversible step would be required to ensure subsequent binding to target MPER epitopes, which are transiently exposed after fusion activation.
Scherer et al. 38 also observed a reduction of lipid binding and neutralization capacities in 4E10 Fabs bearing Trp to Ala substitutions at the H3 loop apex. They concluded that membranotropic H3 loop residues are required to enable MPER recognition through favorable lipid interactions, but cautioned that an alternative option to explain this requirement is that the membrane-bound state may represent the optimal 4E10 epitope configuration.
Ofek et al. 40 focused on the role of the 2F5 H3 loop hydrophobicity on neutralization. Based on mutagenesis of a recombinant 2F5 IgG, they established a quantitative correlation between interfacial hydrophobicity of the loop H3 apex and neutralization capacity, which was independent of the HIV-1 isolate sensitivity to the antibody. They pointed out that 2F5 loop hydrophobicity would effectively promote membrane association only upon proper orientation of this element through epitope binding. To substantiate this prediction, from the water-bilayer partitioning free energies, 29,40 it is possible to estimate the contribution of the loop hydrophobic sequence 100ALFGVPI100F (ideally isolated) to the initial interaction with viral lipid membranes. For a free energy of partitioning, ΔG wiu ≈ −1.5 kcal mol−1, a water-membrane partitioning constant, K x, in the order of 101 can be deduced. Assuming an average of 3 × 105 lipids per viral particle, 53 ca. 1016 virions per ml would be required for half of the 2F5 antibody present in solution to be bound to the viral membrane. This lipid membrane concentration cannot be physically attained for viral particles with diameters of ca. 100 nm, and represents individual molecules separated on average by a distance of ca. 5 Å in an ideal lipid solution. Such a virion concentration can also be compared with the peak plasma viremia during the acute infection phase: in the order of 106 particles per ml of blood. At those virion concentrations, ca. 1 out of 1010 2F5 antibody molecules would be prebound to viral membranes. This figure would increase to ca. 1/108 for the case of the optimized 2F5 versions described by Ofek et al. 40 (i.e., ΔG wiu ≈ −4.5 kcal mol−1, and K x in the order of 103). In conclusion, these estimates strongly argue that the CDR-H3 loop could promote 2F5 antibody membrane insertion only upon specific epitope binding, an event predicted to increase the lipid bilayer effective concentration by orienting and approximating this antibody element toward the viral surface.
Our results in this work further suggest that the CDR-H3 loop insertion into membranes, following linear epitope binding, might play a role in neutralization by interfering with MPER membrane activity. Supporting this hypothesis, only neutralizing 2F5 MAb and Fab wt arrested with comparable potencies leakage induced by the MPER-based 2F5preTM peptide in PG vesicles (Figs. 4 and 5). The experimental data reveal that blocking of the MPER-based peptide activity (Fig. 5), but not leakage directly induced by the antibody (Fig. 3), is an activity dependent on the CDR-H3 loop. Moreover, MPER activity blocking and viral neutralization depend in a comparable fashion on this element (Fig. 6). Thus, our findings establish an unprecedented correlation between MPER membrane-activity blocking by 2F5 and its neutralizing activity.
A common mechanism of action has been proposed for anti-MPER antibodies that could explain 2F5's inhibitory effect on membrane-restructuring activity. According to this mechanism, 2F5, Z13e1 and 4E10 antibodies are capable of inducing large conformational changes in MPER peptides relative to the membrane, which results in partial extraction from the bilayer of several inserted residues. 31,59 Thus, for the particular case of the 2F5 antibody, effective epitope extraction is probably dependent on the association of the hydrophobic loop with the membrane surface. An alternative, nonexclusive possibility is that the loop also establishes secondary interactions with MPER residues located at the C-terminal end of the 2F5 epitope and disrupts the peptide lytic structures. As discussed previously, 41 CDR-H3 apex residues contributed to epitope binding in solution when the residues 672WFNITNWLWYIK683 were added to the gp41 MPER C-terminus.
Interestingly, Xu et al. 39 reported a 4E10 Fv mutant with diminished lipid affinity that otherwise has neutralization potency comparable to or greater than the wild-type version. In view of this evidence, they argue against a “prebinding to viral membrane” model for neutralization by 4E10. They propose a distinct role in the process for the equilibrium immersion depth reached in the lipid bilayer by phosphate-binding subsites and aromatic loop residues. In their model, a shallow insertion of these elements, restricted to the viral membrane interface, might enable the reorientation and pulling out of MPER residues. The observation that the CDR-H3 loop contributes little to lipid monolayer insertion or vesicle leakage (Fig. 3) suggests that this 2F5 element also establishes a shallow interaction with the membrane interface. This loop interaction restricted at the membrane surface level might also be required for effective epitope extraction by the 2F5 antibody. Thus, in-membrane MPER recognition-blocking by 2F5, besides the specific, high-affinity epitope binding, may involve favorable interactions of the loop with lipidic components of virions and/or other regions of the gp41 molecule.
Recovery of ΔCDR3-H3-like responses in rabbits and implications for vaccine design
If a direct antibody–membrane interaction ensues during neutralization, phospholipids might also play a role as additional antigenic targets for the development of effective immunogens. 56,60 The PG membrane model system revealed a correlation between 2F5 Fab neutralizing activity and its capacity for arresting 2F5preTM-induced leakage (Fig. 6). Therefore, we surmised that the 2F5preTM peptide bound to PG vesicles might recapitulate MPER structures relevant for 2F5 neutralization.
Rabbit immunization with 2F5preTM-PG vesicle formulations triggered polyclonal responses specific for the 2F5 epitope (Fig. 7A–C). The purified antibodies were capable of inserting into lipid monolayers and directly inducing PG LUV permeabilization (Fig. 8A and B). Anti-2F5ep antibodies were more efficient at membrane insertion and permeation than the 2F5 antibody. This suggests that a whole range of membrane activities was covered by the polyclonal response, while the lytic activity of the pooled fraction was on average higher than that of 2F5 MAb.
These antibodies were, however, unable to inhibit infection in a pseudovirus assay (Fig. 7D). These findings already emphasize that combining the capacity for binding the peptide epitope sequence with the direct interaction with membranes is not sufficient to recapitulate the neutralizing activity of the 2F5 antibody. Sustaining the correlation existing between neutralization and MPER membrane activity inhibition, the anti-2F5ep antibodies did not block the lytic activity of 2F5preTM assayed in lipid vesicles (Fig. 8C). In summary, at least regarding membrane activity, the polyclonal response recovered in the immunized rabbits is reminiscent of the ΔCDR-H3 Fab mutant. Thus, it is tempting to speculate that the obtained immune response recapitulates on average a 2F5 paratope devoid of the long, hydrophobic CDR-H3 loop. Nonetheless, on the basis of the presented experimental evidence we cannot rule out the existence of other factors, such as insufficient affinity and changes in epitope specificity, that might explain the lack of neutralizing activity of the recovered anti-MPER antibodies.
Alam et al. 61 isolated two MAbs from mice immunized with a gp140 subunit vaccine, which partially cross-blocked 2F5 MAb binding to Env but did not neutralize HIV-1 primary isolates or bind host lipids. They argued that in infected patients B cells could make nonneutralizing cluster II antibodies that may mask, or otherwise down-modulate, B cell responses to regions that could be recognized by B cells capable of producing antibodies such as 2F5. Moreover, MPER-targeting immune responses have been correlated with the production of autoreactive or polyreactive antibodies. 42 Thus, it must be cautioned that the potential application of MPER as a target for vaccination may be hampered by interfering cluster II antibodies and immune tolerance mechanisms.
Finally, our data are also overall in agreement with previous immunization results reported by Alving and co-workers 60,62 showing that multispecific antibodies recognizing both gp41 MPER epitope and lipid can be generated through immunization of mice with liposomal protein. However, they subsequently described the production of neutralizing MAbs with cross-reactivity qualitatively similar to 2F5. 63 Several factors, including differences in the liposome formulation (lipid composition, use of lipid A as an adjuvant), the neutralization assay format, the animal model, or the fact that polyclonal vs. monoclonal responses were assessed, might explain this discrepancy. We speculate that deciphering the basis of these different outcomes would contribute to a better understanding of the factors that regulate 2F5-like neutralizing antibody elicitation. In this regard, molecular and structural analyses of neutralizing vs. nonneutralizing 2F5-based antibodies might provide additional hints for the prospective development of effective anti-MPER immunogens based on liposomal formulations.
Footnotes
Acknowledgments
This study was supported by Spanish MICINN (BIO2008-00772) (J.L.N.), University of the Basque Country (GIU 06/42 and DIPE08/12) (J.L.N.); it was also funded in part by the Ontario Ministry of Health and Long Term Care and by the Canada Research Chairs Program (E.F.P.). R. M. and J.-P.J. were recipients of predoctoral fellowships from the University of the Basque Country and the Canadian Institutes for Health Research, respectively. We gratefully acknowledge the help of A. Cunningham, S. Bryson, and M. Wierzbicka in protein preparation. The views expressed in this publication do not necessarily reflect those of the OMOHLTC.
Author Disclosure Statement
No competing financial interests exist.