
Abstract
Human immunodeficiency virus type 1 (HIV-1) has a high propensity for recombination. The epidemic in South Africa is predominantly driven by HIV-1 subtype C with occasional description of non-subtype C and intersubtype recombinant viruses. This report presents the genetic analysis of a unique recombinant variant from northern South Africa comprised exclusively of subsubtype A1 and subtype C parental viruses. Boot scanning analysis of the near full-length genome with the jumping profile Hidden Markov Model revealed a genomic arrangement with seven breakpoints of recombination alternating between subsubtype A1 and subtype C. Apparently, this is the first report of a unique HIV-1 A1/C recombinant form from northern South Africa and probably the fifth from South Africa. The epidemiologic implication of this variant is unknown.
H
Although subtype C viruses overwhelmingly dominate the epidemic in South Africa, evidence is available on the circulation of non HIV-1 subtype C and recombinant viruses, albeit in low numbers. 6 –9 The vast genetic diversity of HIV isolates and the evolution and spread of novel recombinants pose challenges on the efficacy of diagnostic tools, treatment and treatment monitoring tools, and the evaluation of candidate vaccines. 10 –12 The present study genetically characterizes the near full-length genome of a primary isolate from an individual in northern South Africa, and shows that the isolate has unique recombinant patterns consisting of genomic regions from HIV-1 subsubtype A1 and subtype C parental viruses.
Virus 08BBCR06ZA was obtained from a 33-year-old married female who at the time of blood collection was residing in Bela Bela in the Waterberg District of South Africa. In November 2008 5 ml of venous blood was collected within the context of a genetic drug resistance study. The patient was not on antiretroviral treatment. Her CD4 count and plasma RNA viral load measurements were 38 cells/μl and 60,237 copies/ml, respectively. Ethical approval for the study was obtained from the Health, Safety and Research Ethics Committee of the University of Venda, and permission from the Limpopo Provincial Department of Health.
Viral RNA was isolated from plasma using the QIAmp viral RNA mini kit (Qiagen) according to the manufacturer's instructions. Initial genetic analysis of the complete protease and the partial reverse transcriptase (1296 nucleotides) indicated a subtype assignment of A1 and C, respectively. This observation prompted further investigation of the genomic make up of the isolate since from our previous studies on HIV genetic diversity from about 300 patients based on subgenomic analysis about 99% were pure HIV-1 subtype C with no intersubtype recombinants detected
13,14
(unpublished data). Complementary DNA (cDNA) was generated from plasma RNA, and near-full length viral DNA synthesized by nested PCR as previously described.
15
However, the near-full length nested PCR product was purified and directly sequenced using the 454 Genome Sequencer FLX system as previously described.
16
The subgenomic regions were delineated using the sequence locator software available in the HIV sequence database (
A near full-length genome of 8868 nucleotide long (position 790–9412 relative to HXB2 genome), and spanning the beginning of gag p17 to 3′ LTR, was obtained. Jumping profile Hidden Markov Model analysis showed that the sequence had seven recombination breakpoints alternating between parental subsubtype A1 and subtype C, while REGA analysis showed more than eight recombination breakpoints and unclassified regions (Fig. 1).
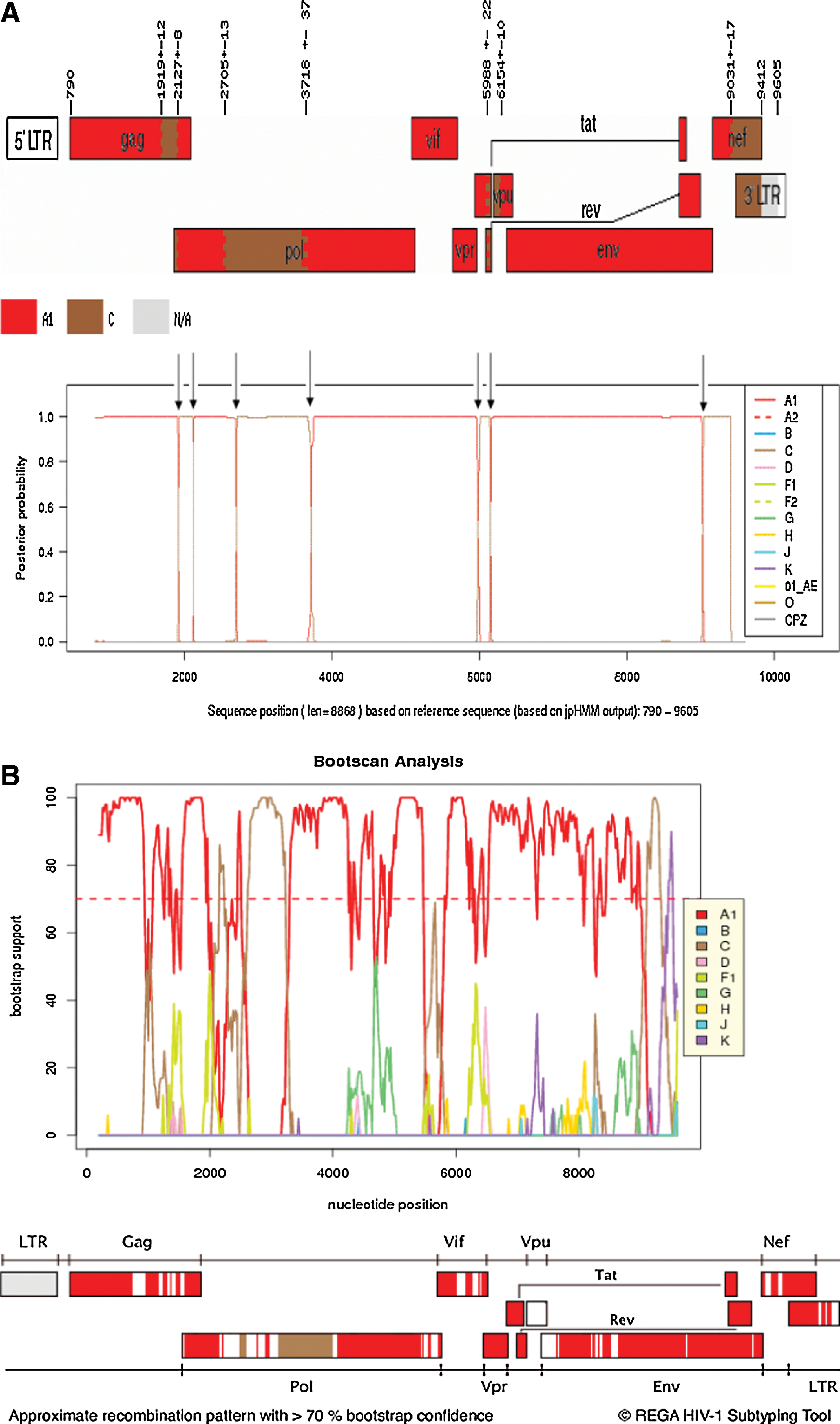
Phylogenetic analysis of the near full-length genome saw 08BBCR06ZA clustering with some A1/C and A1 reference sequences with a bootstrap value of 96% (Fig. 2). However, it should be noted that this clustering is likely influenced by the predominant genome type in the mosaic structure and the reference sequences used. Discrepancies were observed in the assignments obtained with jpHMM, REGA, and phylogenetic analysis when subgenomic regions were considered (see Table 1). The complete gag clustered with sub subtype A1, although p7 was shown to be of subtype C with a bootstrap value of 100%. Similarly, the entire pol gene clustered with subtype C reference sequences with a bootstrap value of 91% even though the protease clustered with subtype A1 references in a separate phylogenetic analysis with a bootstrap value of 87%. Subtyping agreement was seen only in the protease and vpr gene regions (phylogenetic trees not shown). Despite the differences, 08BBCR06ZA is predominantly subsubtype A1 in its genome as revealed by jpHMM, REGA, and phylogenetic analysis.
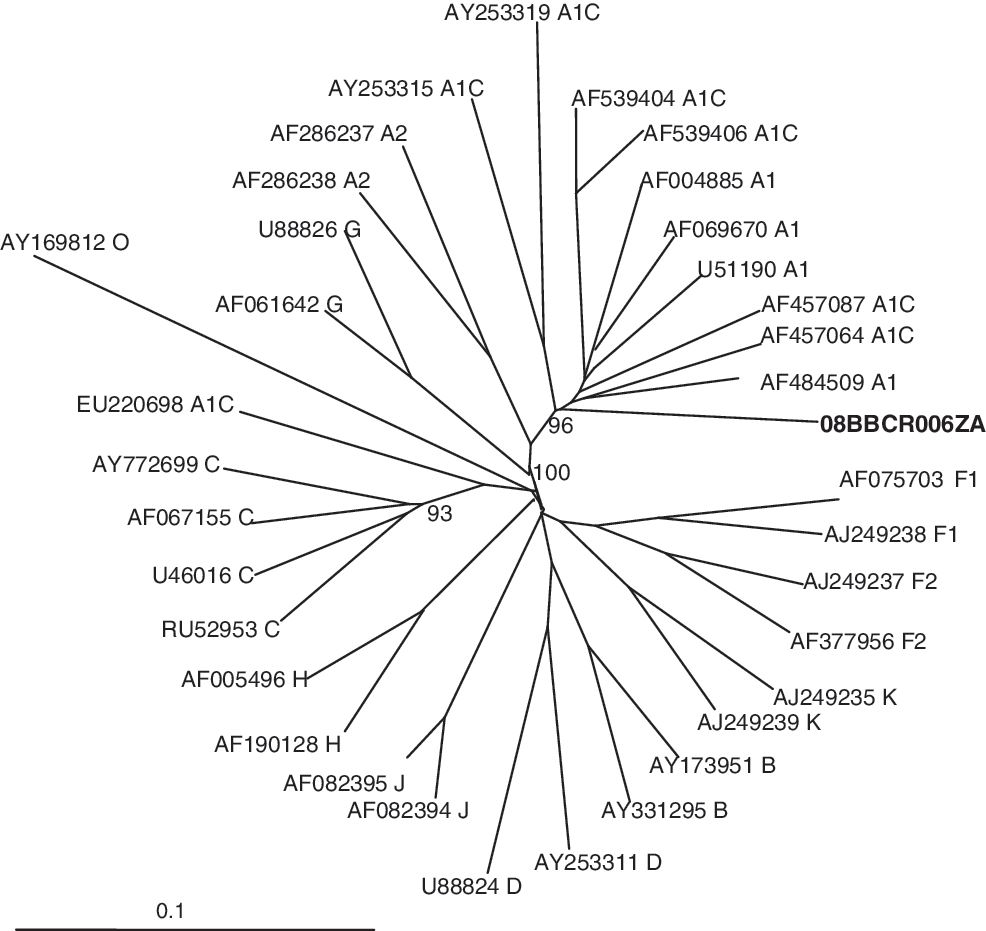
Phylogenetic analysis of the near full-length sequence of 08BBCR06ZA. The test sequence (in bold) is seen clustering with subtype A1 reference sequences and A1/C recombinants with a bootstrap value of 96%. The clustering of A1/C and pure A1 sequences indicates that 08BBCR006ZA is predominately A1 in its genome as evidenced in the bootscanning analyses. The A1/C reference unique recombinant forms obtained from GenBank are of Kenyan, Tanzanian, Rwandan, and Canadian origin. The phylogenetic analysis was done by the neighbor-joining method with 1000 bootstrap resampling, and the tree was visualized with TreeView. Previously described South African A1/C unique recombinant forms were not included in the analysis due to their relatively shorter sequences.
There was complete agreement on subgenomic subtyping by all three methods only for the protease and vpr gene regions. Overall, the genome of isolate 08BBCR06ZA is dominated by subsubtype A1 sequences. U, unclassified gene regions.
Examination of the env gene for biotype prediction revealed conservation of the GPGQ motif with a net charge of +5 in the V3 loop, and webPSSM analysis (
Genetic analyses of HIV is important not only for vaccine development purposes, but also to guide treatment strategies, to track the emergence of new genetic variants, and to ensure that diagnostic assays are continuously able to detect circulating and emerging variants. The HIV-1 epidemic in South Africa is dominated by subtype C, and the prevalence of recombinant viruses seems to be low. Recently, subtypes A1, F1, and a unique A1/C recombinant form were reported from the Cape Town area. 7 Here, we report the isolation of another A1/C recombinant with a profile different from previously reported A1/C variants from South Africa. The virus was isolated from an individual who was infected heterosexually and probably in 2000, and who had never traveled out of South Africa. The presence of an A1/C recombinant in a region where subtype A1 is rare could either mean that the patient had a mixed infection with subtypes A1 and C or was infected by the recombinant virus. Since population-based sequencing was employed in the current analysis it is possible that the A1/C variant was the majority virus in the patient, even though the presence of pure subtypes A1 and C as minority viruses cannot be ruled out. The recombination pattern of isolate 08BBCR06ZA differs extensively in the number of recombination break points and profile from four previously described A1/C variants from South Africa 7,15,19 and from other URF available in GenBank and derived from only subsubtype A1 and subtype C parental viruses.
Extensive analysis of 08BBCR06ZA was prompted when a limited phylogenetic analysis indicated that the complete protease was subtype A1 and the partial reverse transcriptase was subtype C. This underscores the importance of investigating more than one gene region in describing the likely genetic diversity of viruses. South Africa is highly endemic for HIV-1 subtype C, but continuous monitoring of the HIV genetic landscape is important for plausible treatment and prevention implications.
Sequence Data
The nucleotide sequence for 08BBCR06ZA is available from GenBank under accession number GU201611.
Footnotes
Acknowledgments
The South African AIDS Vaccine Initiative, the South African National Department of Health, and the National Research Foundation supported this study. We thank patient 08BBCR006ZA for participating in the study. The views expressed here are those of the authors.
Author Disclosure Statement
No competing financial interests exist.