
Abstract
Human immunodeficiency virus (HIV)-1 targets mononuclear phagocytes (MP), which disseminate infection to organs such as brain, spleen and lymph. Thus MP, which include microglia, tissue macrophages and infiltrating monocyte-derived macrophages (MDM), are important target of anti-HIV-1 drug therapy. Most of the currently used antiretroviral drugs are effective in reducing viral loadin the periphery but cannot effectively eradicate infection from tissue reservoirs such as brain MP. HIV-1 infection of the central nervous system can lead to a wide variety of HIV-1-associated neurocognitive disorders. In this study, we demonstrate that ritonavir-loaded nanoparticles (RNPs) are effective in inhibiting HIV-1 infection of MDM. Reduced infection is observed in multiple read-out systems including reduction of cytopathic effects, HIV-1 p24 protein secretion and production of progeny virions. Furthermore, the RNPs retained antiretroviral efficacy after being removed from MDM cultures. As HIV-1-infected cells in the brain are likely to survive for a long period of time, both acute and chronic infection paradigms were evaluated. Tat-peptide-conjugated RNPs (Tat-RNP) were effective in both short-term and long-term HIV-1-infected MDM. Importantly, we confirm that delivery of RNPs, both with and without Tat-peptide conjugation, is toxic neither to MDM nor to neural cells, which may be bystander targets of the nanoformulations. In conclusion, Tat-NPs could be a safe and effective way of delivering anti-HIV-1 drugs for controlling viral replication occurring within brain MP.
Introduction
T
CNS disease is a common complication of late stage HIV-1 infection that is manifested with moderate to severe neurologic abnormalities (alterations in memory, cognition, motor coordination, and mood) known as HIV-1-associated neurocognitive disorders (HAND). 5,6 Since ART has increased the life expectancy of infected individuals, most believe that the prevalence of HAND will continue to increase. 7,8 HIV-1-associated dementia (HAD), the most severe form of HAND, is associated with productive viral infection of brain MP. 6,9 Clinical disease is often correlated with the neuropathological features of HIV-1 encephalitis, productive infection of brain MP, giant cell formation, monocyte infiltration into the brain, and neocortical atrophy. 10,11 The best histopathologic correlate of HAD is the number of inflammatory macrophages present in the CNS. 12 Therefore, monocyte-derived macrophages (MDM) are not only a target for viral CNS infection but are also an excellent in vitro model for studying brain microglia. While the severity of these symptoms continues to evolve, the brain remains a critical reservoir with significant implications in HAND. To this end, several studies have emphasized the importance of targeting infected tissue MP to achieve better control over the infection and HAND. 2,13 –16
Protease inhibitors (PIs), such as indinavir, saquinavir, and ritonavir, block the viral protease required for HIV-1 assembly and new virion production, and are thus important constituents of ART. 17 Expression of certain efflux transporters, such as P-glycoprotein (P-gp), on the gastrointestinal tract and first path effect contributes toward low oral bioavailability of protease inhibitors. In addition, the high protein binding capacity of PIs reduces the amount of free drug that is available to penetrate tissues and reach the viral reservoir sites. 18 To eradicate viral replication in reservoirs such as brain MP, it is essential that anti-HIV-1 drugs are able to cross tissue and cellular barriers at therapeutic concentrations. Currently, many ARTs are unable to adequately inhibit ongoing viral replication in tissue reserviors. 1,13 The elimination of viral reservoirs is possible only if therapeutic concentrations of antiretroviral drugs are achieved in these tissues and cells over an extended period of time. 19
Colloidal drug delivery systems, such as liposomes, micelles, dendrimers, and polymeric nanoparticles (NPs), have demonstrated increased efficacy in overcoming the many challenges associated with current antiretroviral drugs, thus improving the overall management of anti-HIV-1 therapy. 20 NPs are sustained release delivery systems in which, the therapeutic agent can be encapsulated. Further, NPs can be endocytosed by cells leading to better drug delivery than drugs in solution, with a concomitant reduction in side effects. The delivery of drug-encapsulated NPs can be further enhanced by conjugating to specific targeting agents such as cell-penetrating peptides (CPPs). CPPs, such as the transactivator of transcription (Tat)-peptide, containing a sequence of highly basic amino acids, can interact with the cell surface via a receptor-independent mechanism and can transport NPs attached to them across the cell membrane. 21 The mechanism of cellular entry of Tat-peptide and the attached encapsulated drug is shown to be independent of the cell type; several studies have demonstrated increased transport and uptake of Tat-peptide-conjugated cargo across numerous cell lines. 22 –24 Recently, we have demonstrated significantly higher bioavailability of ritonavir in the brain following treatment with Tat-RNPs than nonconjugated ritonavir-loaded nanoparticles (RNPs) or nonencapsulated ritonavir. Tat-peptide conjugation was also shown to increase the uptake of NPs within macrophage-rich organs such as lungs, liver, and spleen. 25
Since the NPs are capable of crossing the blood–brain barrier (BBB), and are available in the brain to be engulfed by MP, they are also in proximity of brain neurons. Although it is generally accepted that neurons do not become actively infected by HIV-1 and thus are not the intended targets of ART, neurons may be particularly sensitive to NPs, their degradation, and subsequent release of encapsulated drug. Furthermore, Tat-peptide conjugation used to enhance NP delivery to resident CNS MP may cause unintended neurotoxicity, as full-length Tat protein is a known proapoptotic viral protein in its full-length form. 26 –28 Therefore, the neurotoxicity of the NP, CPP, and drug delivered should be determined prior to suggesting this delivery mechanism to eradicate HIV-1 reservoirs in the brain.
In this study, we determined the efficacy of drug delivery to MDM in vitro, and possible toxicity and cytopathicity of NPs to MDM. Furthermore, we determined if Tat-NP could deliver an effective dose of drug to MDM to inhibit viral replication during both acute and chronic HIV-1 infection, and if Tat-peptide conjugation would lead to increased toxicity in MDM. Finally, we evaluated if NP, Tat-peptide conjugation, or the delivery of drug would have adverse side effects on cultured primary human neurons. These studies would help elucidate the drug delivery efficacy of Tat-NPs, and if they are a biocompatible choice for future ART CNS drug delivery.
Materials and Methods
Formulation, conjugation of Tat-peptide, particle size analysis, and zeta potential of NPs
NPs were formulated using an emulsion-solvent evaporation technique as previously described. 25 Tat-peptide of the sequence Tyr-Gly-Arg-(Lys)2-(Arg)2-Gln-(Arg)3 (molecular weight 1917 Da) was custom synthesized by Invitrogen Corporation (Carlsbad, CA). Tat-peptide was conjugated to RNPs and CNPs by an epoxy conjugation method and quantified as previously described. 25 Denacol EX-521 (Pentaepoxy, molecular weight 742 Da), an epoxy-linker used to activate the NP surface, was a gift from Nagase Chemicals Ltd (Tokyo, Japan). The mean hydrodynamic diameter and zeta potential of RNPs, Tat-RNPs, and their respective controls were measured using a ZetaPlus particle size analyzer (Brookhaven Instrument Corp., Holtsville, NY). The mean hydrodynamic size of nonconjugated NPs was 285 nm (Polydispersity Index, PI = 0.103), while that of Tat-NPs was 314 nm (PI = 0.197). The zeta potential of nonconjugated RNPs was −22.0 ± 0.4 mV, which changed to a slightly positive value of +2.64 ± 0.2 mV after Tat-peptide conjugation. From our earlier studies, it was found that loading of ritonavir in NPs was 18.3% (w/w), 80% of the drug was released from the NPs in 10 days, and the amount of Tat-peptide bound was 0.23 μg/mg of NPs. 25
The concentration of nonencapsulated ritonavir used in vitro is based on the plasma ritonavir concentration (∼1.6 μM), which is effective in reducing the viral load in blood in HIV-1-infected patients 29 and release rate of the encapsulated drug from NPs (∼80% in 10 days). 25 The concentration of nonencapsulated ritonavir is steady throughout all experiments. However, the concentration of NP was adjusted to accommodate the breakdown of the NP via hydrolytic cleavage of polymer chains over various incubation periods. NP concentrations expressed as NP equivalent to molarity of ritonavir.
Isolation and culture of monocyte derived macrophages (MDM)
Monocytes were purchased through the Tissue and Cells Core Facility, Center for Neurovirology and Neurodegenerative Disorders, University of Nebraska Medical Center, Omaha, NE. MDM were cultured as previously described. 30
HIV-1 infection of MDM
MDM were cultured on 48-well plates (Costar, Cambridge, MA) at a seeding density of (5 × 104) cells per well. After 7 days in culture, MDM were infected with HIV-1ADA (a macrophage tropic strain) at a multiplicity of infection (MOI) of 0.01 infectious viral particles/target cell as described previously. 30 One day postinfection, the medium used for infection was exchanged with fresh MDM media.
Antiretroviral activity in MDM
The antiretroviral activity of NPs was determined in infected MDM by assessment of (1) reverse transcriptase (RT) activity, (2) Cytopathicity, and (3) HIV-1 p24 antigen level.
RT activity
Viral RT levels in the culture were measured as one indicator of the anti-HIV-1 activity of different formulations as described previously. 31 MDM cells were cultured in 48-well plates in triplicate conditions. The cells were infected with HIV-1ADA after 1 week in culture. Two days postinfection the cells were exposed to nonencapsulated ritonavir and drug loaded in NPs at 12.5 μM. Ritonavir drug stock solution was prepared in ethanol (to dissolve ritonavir) and subsequently diluted with culture medium to 10 μM, which was earlier found to be effective in solution form in reducing HIV-1 replication in infected MP. 32 NPs were dispersed in DMEM by sonication in a water bath for 1 min. Appropriate dilutions of NP suspension were prepared in serum containing DMEM prior to the experiment. The final volume in each well was 0.5 ml. Following treatment, one set of cells was left unwashed in which NP or nonencapsulated ritonavir was present throughout the duration of the experiment (No Wash). In the second set of cells, NP treatment was removed after 48 h (Wash) by two full media exchanges. The Wash and No Wash treatments were carried out to determine the stability of the NP treatments as compared to No Wash nonencapsulated ritonavir and untreated HIV-1-infected control. MDM culture supernatants were collected on day 7, or 5 days postwash, and the virion production was assessed by measuring RT activity. RT activity was determined in triplicate.
Cytopathicity
MDM were fixed with 1:1 acetone:methanol for 15 min at −20°C and immunostained for CD68 (macrophage marker, green) to visualize the changes in cytopathicity of MDM cells due to infection and NP treatment under a fluorescent microscope (200 × original magnification, Nikon Microphot-FXA microscope, Nikon Inc., Japan).
HIV-1 p24 assay in acute infection
To study the inhibition of HIV-1 infection, 2 days postinfection of MDM with HIV-1ADA, cells were exposed to nonencapsulated ritonavir or Tat-NPs (10 μM) for 48 h. On the seventh day postinfection, or 5 days postwash, culture supernatants were collected and assayed to quantify HIV-1 p24 levels by ELISA (PerkinElmer Inc., Waltham, MA) as per the manufacturer's directions. All experiments were performed in triplicate.
HIV-1 p24 assay in chronic infection
To obtain chronic infection, MDM were infected with HIV-1ADA for 3 months. During this time, all cells received a 50% media exchange twice a week. At the end of this period, the growth medium was completely replaced with fresh medium for 24 h providing a starting infection level for each treatment group (d0). MDMs were then treated with nonencapsulated ritonavir (10 μM), Tat-CNPs (10 μM), Tat-RNPs (10 μM), and nonconjugated, truncated Tat-peptide (50 μg/ml). Untreated-infected cells were maintained in parallel. Supernatant samples from each treatment group every 24 h for 1 week posttreatment and HIV-1 p24 levels were quantified by ELISA. The HIV-1 p24 levels in each treatment group were normalized to their respective d0 HIV-1 p24 levels as percent d0 for all time points. Untreated HIV-infected cells were used as a control for progressive chronic infection and nonencapsulated ritonavir for active antiretroviral activity.
Effect of NPs on the viability of MDM cells
The effect of NPs and nonencapsulated ritonavir on the viability of MDM cells was determined colorimetrically using MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (Sigma). The amount of MTT formazan produced is indicative of cellular viability. This test was performed on noninfected MDM treated with nonencapsulated ritonavir, CNPs, RNPs, Tat-CNPs, Tat-RNPs, and nonconjugated truncated Tat-peptide, according to the manufacturer's instructions.
Isolation and cultivation of cultured human neurons
Human fetal brain tissue was obtained from abortus materials that were gathered by the Birth Defects Research Laboratory at University of Washington (Seattle, WA). All the studies were carried out in accordance with approved guidelines of the National Institutes of Health (NIH), the Universities of Nebraska Medical Center (UNMC), North Texas Health Science Center (UNTHSC), and Washington (UW). The Birth Defects Laboratory has been continuously funded by the NIH for >40 years for the collection of tissues for research and is compliant with the relevant State and Federal Regulations. The obtained tissue was transported in Hanks' balanced salt solution (HBSS) at 4°C prior to isolation of neuronal cells. Human neuronal cultures were prepared using the protocol as previously described. 33
Viability of human neurons upon exposure to NPs
Cultured human neurons were half exchanged with neuron media containing nonencapsulated ritonavir, NPs, nonconjugated truncated Tat-peptide, and staurosporine (STS). Untreated control cells were half-exchanged with neuronal medium. The final concentration of all NPs, both Tat-peptide-conjugated and not, was 10 μM; nonencapsulated ritonavir was 10 μM and nonconjugated, truncated Tat-peptide was 50 μg/ml. STS used as a positive control for reduced viability was used at 1 μM. The drug and NP stocks were prepared as discussed above. Neuronal toxicity was assessed by two methods, namely apoptosis as measured by double-stranded DNA fragmentation ELISA (Roche Applied Sciences, Indianapolis, IN) and cell death or cell lysis as measured by the lactate dehydrogenase (LDH) assay (Roche) according to the manufacturer's instructions. All the experiments were performed in triplicate in three neuron donors.
Statistical analyses
All experiments were performed in a minimum of three replicates in multiple donors. Statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA). Differences between groups were analyzed by one-way ANOVA and the Newman–Keul's multiple comparisons test was used for post-hoc analysis.
Results
Effect of NPs on MDM viability
Prior to assessing the effect of the NP formulations on HIV-1 infection, possible adverse cytotoxic effects of NPs were assayed. Thus, uninfected MDM were treated with NPs (10 μM, CNPs and RNP) and viability was measured by MTT assay. All samples were normalized to the metabolic activity of noninfected, control untreated MDM, and were plotted as percent control. During 9 days of treatment, neither CNPs nor RNPs demonstrated cytotoxicity as compared to control (Fig. 1A). Nonencapsulated ritonavir, used as a control for verifying direct effects of the drug, also did not significantly affect MDM viability (Fig. 1A). Noninfected MDM were used to assay the cytotoxicity of treatments rather than infected MDM because viral infection would skew the results toward the antiretroviral treatments. Thus, phagocytosis and degradation of CNPs and RNPs do not affect MDM viability. Furthermore, ritonavir whether encapsulated or not is also nontoxic to MDM.
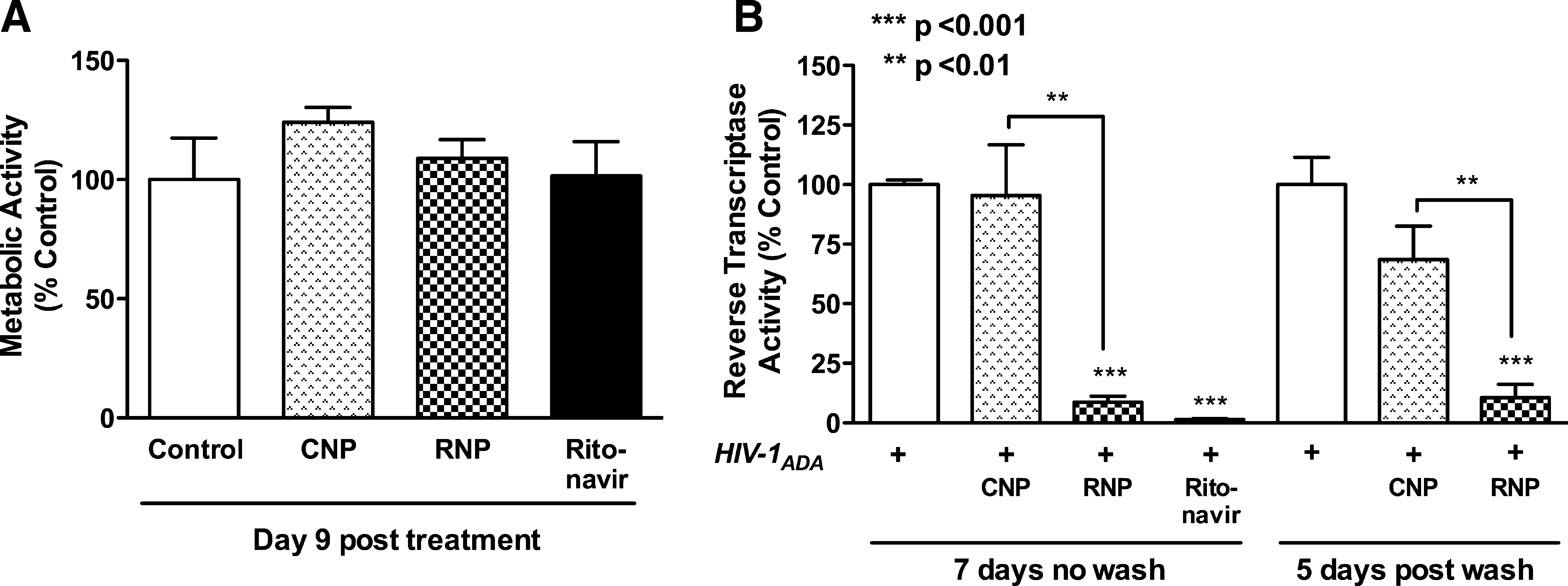
Nanoparticle (NP) cytotoxicity and antiretroviral activity.
Stability and antiretroviral activity of RNPs in HIV-1-infected MDM
Having established that NPs are not cytotoxic to MDM, the antiretroviral efficacy of nonencapsulated ritonavir and RNPs was evaluated by measuring the RT levels in MDM culture supernatants in two treatment paradigms: Wash, 48 h NP (12.5 μM) treatment followed by a full media exchange and No Wash, 7 days NP treatment with no media exchange. Again, all samples were normalized to the metabolic activity of Wash or No Wash infected control MDM and were plotted as percent respective controls. Nonencapsulated ritonavir (10 μM) treatment was constant, without wash, as a positive control for antiretroviral activity. As expected, the No Wash treatment paradigm showed significant reductions in RT activity upon treatment with RNPs as compared to both No Wash control and CNP-treated MDM 7 days posttreatment (Fig. 1B, p < 0.001 and p < 0.01, respectively). Thus, the MDM were able to phagocytose the NPs and release the encapsulated ritonavir, which retained its antiretroviral activity. Furthermore, washed cells also showed a significant 68% reduction in RT activity as compared to washed untreated control and CNPs (Fig. 1B, p < 0.001 and p < 0.01, respectively) 5 days postwash or 7 days posttreatment. Thus, the ritonavir encapsulated in the phagocytosed NPs was released over time and was equally effective as No Wash treatment RNP. Both RNP treatment paradigms resulted in higher RT activity levels than nonencapsulated ritonavir; however, neither was significant (Fig. 1B, p > 0.05).
Cytopathicity in MDM during HIV-1 infection and NP treatment
A morphological hallmark and measure of HIV-1 infection in cultured MDM is the formation of multinucleated giant cells (MGCs) due to the cytopathic effects of HIV-1. To evaluate the changes in the levels of cytopathicity, MDM were infected with HIV-1 for 2 days and then treated with NPs (12.5 μM) and nonencapsulated ritonavir (10 μM) for 7 days. Cells were fixed with acetone:methanol (1:1) and stained with macrophage marker CD68 (green) and nuclear marker DAPI (blue). HIV-1 infection of MDM induced formation of characteristic MGCs (Fig. 2A) as compared to noninfected controls (Fig. 2A, inset). Treatment with nonencapsulated ritonavir and RNPs inhibited the development of MGCs, reducing viral infection and cytopathicity (Fig. 2C and D), while treatment with CNPs showed little effect (Fig. 2B). These effects mirrored those measured by RT activity (Fig. 1B).
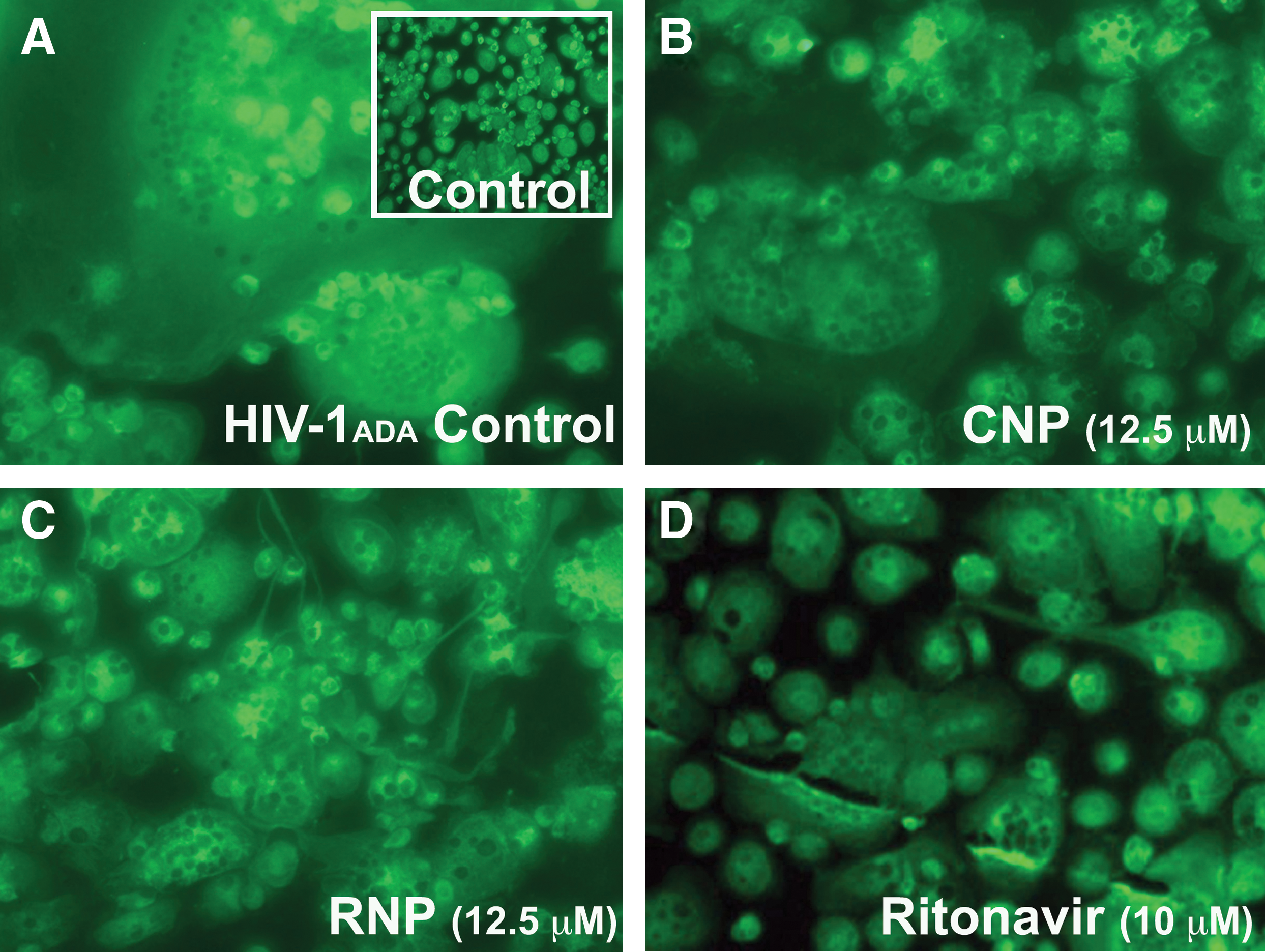
Cytopathicity of HIV-1 infection following treatment with NPs. MDM were infected with HIV-1 for 2 days
Tat-NP stability and efficacy in infected MDM
Since RNPs could reduce infection levels in both constant and wash models without cytotoxicity, we then investigated the effect of Tat-peptide conjugation to the NP. We have previously shown that Tat-peptide conjugation to NP can increase the bioavailability of drug in the brain. 25 However, we wanted to determine the antiviral efficacy and toxicity of NP conjugated to a small segment of the Tat protein in acute and chronic versions of our culture model.
In an acute 7-day infection, similar to that used to assess the antiretroviral activity of RNP, the effects of Tat conjugation on NPs were assessed in infected MDM. Two days postinfection, MDM were treated for 48 h with Tat-NPs (10 μM). Cultures were then washed to remove nonengulfed NPs and incubated for 3 days prior to collection of cellular supernatants. Infected MDM were continuously treated with nonencapsulated ritonavir (10 μM) as a positive control for antiretroviral effects. Since RT activity measures only progeny virions produced by infected cells, we chose to quantify HIV-1 p24 production, as all infected cells secrete HIV-1 p24 during infection. The HIV-1 p24 ELISA can detect levels as low as 17.1 pg/ml and thus increases the sensitivity for detecting changes in MDM infection. Cellular supernatants collected 7 days postinfection or 5 days posttreatment were assayed. Tat-RNPs significantly reduced HIV-1 p24 levels by 70 % (p < 0.01) as compared to both control and Tat-CNPs MDM (Fig. 3A). Tat-RNP treatment at 5 μM was also effective at reducing HIV-1 p24 levels under these experimental conditions (data not shown). Non encapsulated ritonavir also significantly reduced HIV-1 infection by 80% (p < 0.001); however, it was not significantly less than Tat-RNP.
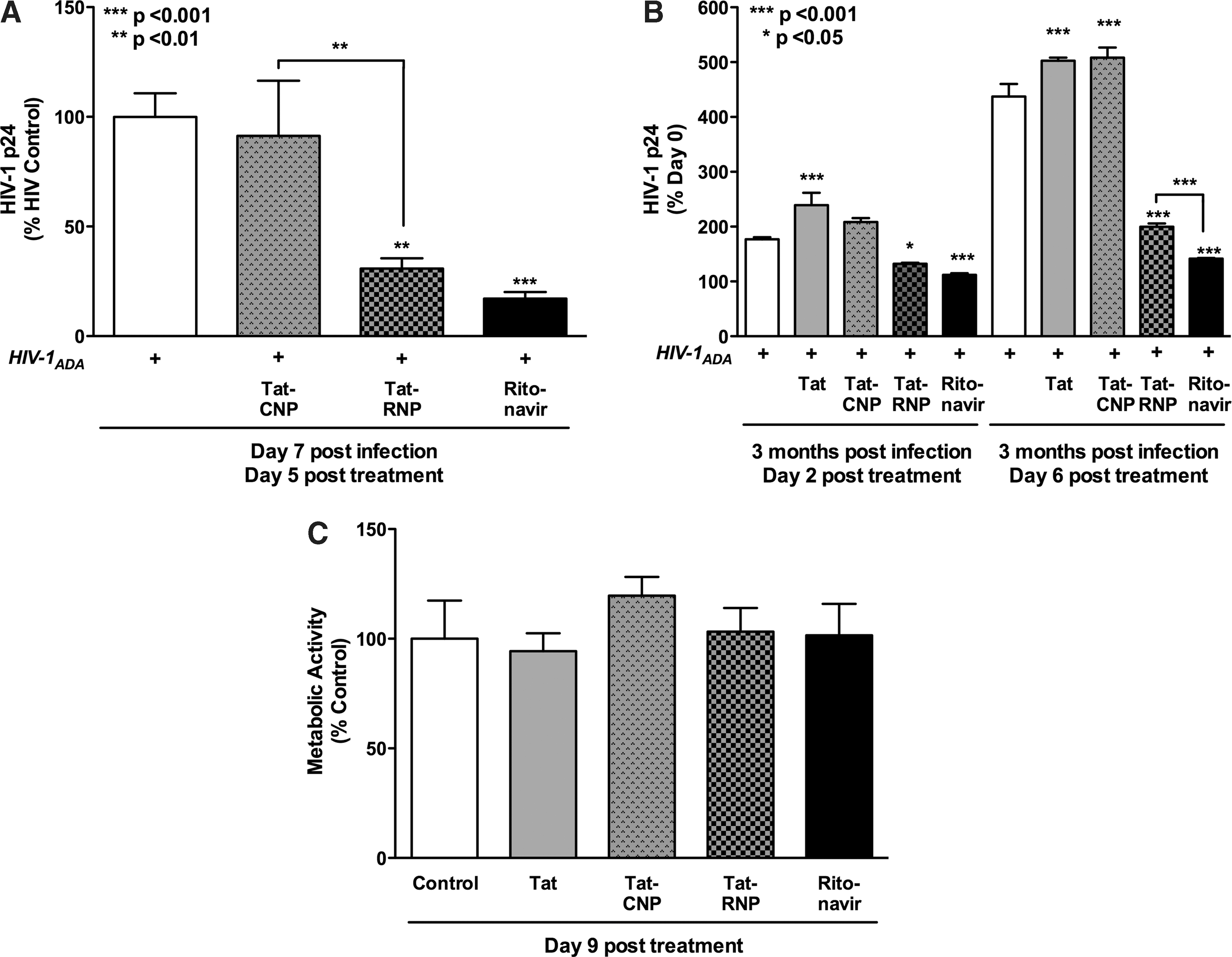
Tat-NP antiretroviral activity and cytotoxicity.
In a separate experiment, MDM were chronically infected with HIV-1 for 3 months. HIV-1 p24 levels in culture supernatants were assayed by ELISA prior to NP treatment to establish a d0 baseline infection level. Infected MDM were treated with Tat-NPs (10 μM) for 6 days without removing the NPs or media exchanges. The MDM were also treated with nonconjugated Tat-peptide (50 μg/ml) and nonencapsulated ritonavir (10 μM). Supernatant aliquots were collected 2 and 6 days posttreatment and assayed for cumulative HIV-1 p24 levels as a measure of productive infection. On day 2, both Tat-RNPs (Fig. 3B, p < 0.05) and nonencapsulated ritonavir (p < 0.001) reduced HIV-1 p24 levels by 75% and 63% that of untreated controls. However, the effect was much more dramatic in day 6 samples with HIV-1 p24 levels less than 50% of control (p < 0.001). In nonconjugated Tat-peptide and Tat-CNP treated MDM, infection levels increased at both day 2 and day 6 due to lack of antiretroviral treatment. The ability of Tat-RNPs to release ritonavir and decrease active viral replication in MDM was pronounced with over 2-fold less infection than control by day 6.
Effect of Tat-NPs on the viability of MDM cells
We assessed the cytotoxicity of the Tat-NPs considering that full-length Tat is an activating protein, likely to affect cells in high doses. During 9 days of treatment, neither 10 μM Tat-CNPs nor Tat-RNPs demonstrated cytotoxicity as compared to control in noninfected MDM as measured by the MTT metabolic assay. Furthermore, nonencapsulated ritonavir (10 μM) and nonconjugated Tat-peptide (50 μg/ml) also did not significantly alter MDM viability (Fig. 3C, p > 0.05). Thus, Tat-peptide-conjugated NP formulations were biocompatible with human MDM.
Neurotoxicity of NPs
Although neurons are not phagocytotic cells, NPs or CPPs could affect neurons through interactions in the intercellular milieu after crossing the BBB. Thus, to ascertain the effect of NPs on human neurons, cultures were treated for 24 h with both Tat-peptide-conjugated and nonconjugated NPs (10 μM), nonencapsulated ritonavir (10 μM), and the truncated Tat-peptide used for conjugation (50 μg/ml). Cultures were also treated with staurosporine (STS, 1 μM) as a positive control for cytotoxicity. Neurons treated with CNPs, RNPs, Tat-CNPs, Tat-RNPs, truncated Tat-peptide, and nonencapsulated ritonavir did not show any significant differences in apoptosis levels as compared to untreated controls (Fig. 4A, p > 0.05). As expected, STS significantly increased apoptosis by 1.8-fold (p < 0.001). In a parallel assay on the culture supernatants, human neurons showed no increase in LDH release for any NP treatments as compared to untreated control (Fig. 4B, p > 0.05). STS, the positive control, showed a 5.85-fold increase over that of untreated control (p < 0.001). Thus, all forms of NP, both Tat-peptide-conjugated and unconjugated, or ritonavir loaded and empty, did not affect the viability of human neurons. Furthermore, nonencapsulated ritonavir and nonconjugated, truncated Tat-peptide were not directly cytotoxic to human neurons.

Neither NPs nor Tat-NPs are neurotoxic. Human neurons were treated with NPs (10 μM), Tat-NPs (10 μM), nonconjugated truncated Tat-peptide (50 μg/ml), nonencapsulated ritonavir (10 μM), and staurosporine (STS, 1 μM, positive control) for 24 h.
Discussion
In this study, we demonstrate that RNPs are effective in inhibiting HIV-1 infection of MDM. Reduced infection is observed in multiple read-out systems including reduction of cytopathic effects, HIV-1 p24 protein secretion, and production of progeny virions. Indeed, as brain is a critical reservoir for virus and brain MP harbor HIV-1 for a long duration, Tat-peptide conjugation of RNPs was used as a strategy for effective targeting to the brain in earlier studies. 25 As HIV-1-infected cells in the brain are likely to survive for a long period of time, both acute and chronic infection paradigms were evaluated. Tat-RNPs were effective in both short-term and long-term HIV-1-infected MDM. In this study, we confirm that delivery of Tat-RNP is toxic neither to MDM nor to neural cells, which may be bystander targets of the nanoformulations.
Recently, studies of HIV-1-infected patients on PI-based ART have shown various side effects from treatment referred to as the “cardiometabolic syndrome.” These side effects include peripheral lipodistrophy, visceral adiposity, hyperlipidemia, insulin resistance, hyperglycemia, and overt type two diabetes mellitus. 34,35 In a study evaluating skeletal muscle fatty acid oxidation, ritonavir was used as the stable component of the ART therapy while studying the effects of atazanavir, lopinavir, and darunavir. 36 Ritonavir exhibits limited cellular permeability, attributed mainly to its efflux by the P-gp transporter and to extensive protein binding, resulting in lower intracellular drug levels. 37 Ritonavir continues to be used in clinical pharmacokinetic studies for drug combinations, safety and side effects in healthy noninfected patients, pregnant women, children, and in patients with HIV-1 drug resistance. 38 –47 ART including ritonavir remains a viable option for HIV-1 treatment; therefore, it is relevant to evaluate its efficacy and possible adverse side effects following NP delivery to the brain.
The goal of any drug delivery system is to exhibit maximal efficacy without unwarranted toxicity. Several studies have emphasized the biocompatibility of the polymers, polylactide and its glycolide copolymer, used in our studies for formulating NPs across various cell lines and in vivo models. 48 Our earlier studies have demonstrated enhanced uptake of ritonavir with Tat-RNPs as compared to nonencapsulated ritonavir by P-gp expressing MDCK-MDR cells. 25 However, it is pertinent to determine the biocompatibility of these NPs in human cells for clinical applications. To assess NPs as a drug delivery system to brain cells, direct NP toxicity to the target MP cells and nontargeted human neurons was determined. Neither nonconjugated nor Tat-NP showed direct toxicity or adverse effects on cellular morphology of MDM following continuous treatment for 9 days. Furthermore, NP formulations were not directly neurotoxic in vitro. Previously, Reddy et al. demonstrated the neuronal biocompatibility of PLGA-NPs even at a dose of 1 mg/ml for 24 h. 49
Of greater possible concern is the known neurotoxicity of the full-length Tat protein, 26 of which a portion was used to promote transfer of NP across the BBB. In neurons treated with high concentrations of full-length Tat, a generalized dysregulation of calcium has been observed, which leads to NO production and apoptosis. Tat has also been found to alter microtubule dynamics in human 293T and Jurkat cells leading to apoptosis. 26 However, severe neuronal damage in mice was observed only in the case of supraphysiological concentrations of Tat-peptide (13.5 μg–180 μg). 27 Earlier studies have demonstrated that only nanogram levels of the peptide were sufficient to deliver therapeutic levels of ritonavir-loaded within Tat-NPs to the brain. 25 During our in vitro assessments, Tat-peptide conjugation did not increase NP neurotoxicity. The Tat-peptide used for conjugation is not the full-length protein, but only a small segment of the peptide that increased NP delivery to the brain in previous studies. 25 The truncated Tat-peptide used in this study does not promote HIV-1 replication in monocytes, 25,50 and is nonneurotoxic at over five times the amount conjugated to an effective dose of Tat-RNP. Thus, even as the Tat-peptide is released from the NP upon degradation, the levels are biocompatible, as shown by the lack of toxicity in Tat-NP treatments in both MDM and neurons. Tat-NP may therefore be a viable option for therapeutic delivery of ART to the CNS.
After confirming the lack of cellular toxicity, we evaluated the efficacy of the NP formulations to effectively inhibit viral replication in acute and chronic HIV-1 infections. HIV-1-infected individuals seek treatment at various stages of disease progression. Additionally, HIV-1 can infect the brain and become a reservoir for the virus early in infection, even when viral infection is adequately treated in the periphery. 4 It is therefore essential to control the ongoing viral replication in these cells to ensure complete eradication of the virus. 51 ART should be effective even when therapy is initiated after a chronic infection has been established. Poor efficacy of PIs against chronically infected macrophages remains one of the major drawbacks of the current ART. 8,52 Viral antigen production increases immediately after halting drug therapy from chronically infected cells that carry infectious DNA integrated within the host genome rendering the treatment ineffective. 52 In our study, both nonconjugated and Tat-RNP effectively reduced HIV-1 infection during 7 day acute infections in MDM. When the NP were washed away and further NP treatment was discontinued, viral levels failed to increase for 5 days. This finding clearly suggests that NP sustain drug availability at levels capable of inhibiting productive viral replication without toxicity in vitro for an extended period of time. RNP antiretroviral activity was evaluated across multiple parameters. Reduced progeny virion levels correlated with decreased cytopathic effects, and treatment with Tat-RNP led to a reduction in HIV-1 p24 secretion. Tat-RNP effects were also evaluated in a chronic timeframe (3 months postinfection). Significantly lower HIV-1 p24 levels were observed 2 and 6 days posttreatment, as compared to no ritonavir controls.
Recently, other studies have also demonstrated the effectiveness of nanotechnology loaded with combinations of antiretroviral drugs in reducing viral levels during both acute and chronic stages of HIV-1 infection. 16,53 –56 For example, Bender et al. demonstrated that in 1 month chronically infected MDM cells, the viral antigen production reduced by 35% in the saquinavir-loaded NP group, whereas no inhibitory effect was observed in the solution group. 57 Nowacek et al. investigated the efficacy of nanocrystals of different antiretroviral drugs, which were coated either with surfactants, lipids, or encapsulated in a polymer (NanoART) to inhibit the release of progeny virion from virus-infected MDM in vitro. 15,58 The effect of different formulations of NanoART varied but was dose dependent and sustained.
From our previous studies, it is estimated that the approximate concentration of ritonavir released from either type of NP in 48 h would be 0.48 μM ritonavir/μM NP (based on 50% drug release in two days in vitro). 25 Based on these in vitro studies in infected macrophages, a 10 μM dose of ritonavir encapsulated in Tat-NPs effectively inhibited viral replication over 7 days. While extrapolating a human dose from in vitro studies is difficult, the Tat-RNP dose necessary to suppress viral replication in humans could be ∼19 g, i.e., 3.6 g of encapsulated ritonavir. This estimation is based on ∼5 liters total blood volume and 18.3% ritonavir loading in NPs. This dose is feasible for injection as an intravenous infusion of NP suspension.
Overall, by encapsulating antiretroviral drugs within NPs and Tat-NPs, drug delivery to inaccessible cells in the brain and other tissues can be improved. We specifically formulated polymeric nanoparticles loaded with ritonavir and conjugated to a Tat-peptide. In previous studies, 25 we have shown that Tat-peptide-conjugated NPs can pass the BBB and transport the encapsulated drug to the brain parenchyma without disrupting the integrity of the BBB. Furthermore, therapeutic drug levels in the brain were maintained over 2 weeks with a single-dose intravenous injection of NPs in mice. The sustained release properties and enhancement in drug bioavailability and biocompatibility make these Tat-peptide-conjugated NP formulations safe and effective to warrant testing in disease conditions. Thus, Tat-peptide-conjugated NPs can potentially target macrophages in the periphery as well as the CNS and perhaps other organs that may serve as viral reservoirs.
Footnotes
Acknowledgments
This study was supported by NIH Grants 1R01 NS048837-06 (to A.G.) and 1R21 MH67525 (to V.L.).
Author Disclosure Statement
No competing financial interests exist.